
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Aperçu du séquençage de l'ADN
Technologie de séquençage génomiqueégalement appelé Technologie de séquençage de l'ADN, implique l'acquisition de l'agencement des nucléotides d'un segment d'ADN ciblé. Dans le domaine de la recherche scientifique, obtenir la séquence d'un fragment d'ADN cible constitue l'étape fondamentale pour les investigations ultérieures en biologie moléculaire et les modifications génétiques. Alors que le paysage de la génomique, particulièrement propulsé par des avancées telles que Études d'association à l'échelle du génome (GWAS), continue de mûrir, l'anticipation du risque de maladie et le diagnostic précoce grâce aux approches génomiques ont émergé comme une trajectoire essentielle en médecine clinique. De plus, la médecine de précision, saluée comme une voie clé pour l'avenir de la médecine clinique, souligne l'importance indéniable de la technologie de séquençage génomique dans son paradigme. Chaque évolution de la technologie de séquençage représente un impetus monumental pour divers domaines, y compris la recherche génomique, les thérapies contre les maladies et le développement pharmaceutique. Ces avancées transformantes contribuent continuellement à l'accélération et à l'enrichissement de notre compréhension et de nos capacités en génomique, façonnant finalement le paysage de la recherche et de la pratique médicales.
Développement de la technologie de séquençage
L'évolution de la technologie de séquençage de l'ADN a été remarquable depuis son origine avec la méthode Sanger de première génération. Basé sur des principes différents, le progrès de la technologie de séquençage peut être segmenté en trois étapes : première génération Séquençage de Sangerdeuxième génération séquençage à haut débitet de troisième génération monomoléculaire/séquençage par nanoporeChaque génération a ses propres forces et faiblesses ; par conséquent, leurs applications varient en conséquence. Actuellement, le marché du séquençage est dominé par Séquençage de nouvelle génération (NGS), mais il permet également la coexistence du séquençage à longues lectures. Séquençage à lecture longue technologies.
Service qui pourrait vous intéresser
Séquençage de Sanger
les technologies de séquençage de première génération comprenaient la méthode de terminaison par double désoxy-nucléotides de Sanger et la méthode de dégradation chimique innovée par Alan Maxam et Walter Gilbert. Cette technologie révolutionnaire a facilité le séquençage automatisé des molécules d'acides nucléiques. En 1977, le séquençage de première génération pouvait atteindre une longueur de séquençage de 1 000 paires de bases (pb) avec un taux de précision élevé de 99,99 %, fournissant un outil avancé pour la recherche en biologie moléculaire. Pendant une période considérable, cette méthode était très appréciée pour ses longues lectures, sa grande sensibilité et sa précision, jouant un rôle crucial dans le projet génome humain précoce. Cependant, Séquençage de Sanger a ses limites ; bien que la technique fournisse des lectures plus longues par rapport au séquençage de deuxième génération ultérieur, chaque course de séquençage ne peut produire qu'une séquence d'environ 700 à 1000 paires de bases de longueur, car la longueur de chaque lecture est la limite de sa capacité de séquençage en une seule course. Étant donné qu'une conception de primer distincte est requise pour chaque séquençage, face à la demande généralisée de séquençage à l'échelle du génome dans la recherche moderne et les applications cliniques, le débit relativement faible, le processus long et les coûts plus élevés du séquençage Sanger ont limité ses perspectives pour des applications à grande échelle.
Progressivement, la dominance du séquençage de première génération a été remplacée par le séquençage de deuxième génération émergent (séquençage de nouvelle génération ou NGS). Cependant, la technologie de séquençage de première génération possède encore certains avantages, tels qu'une grande précision, des lectures plus longues et l'élimination de la nécessité d'assembler les séquences. Par conséquent, elle conserve une certaine importance dans certains domaines de recherche. Par exemple, elle joue un rôle essentiel dans des domaines tels que le séquençage de plasmides, la validation de sites de mutation, le séquençage de petits génomes bactériens, le séquençage des extrémités de chromosomes artificiels bactériens et le séquençage de séquences répétitives.
 Figure 1 Principe du séquençage de Sanger (Michel G Gauthier 2007)
Figure 1 Principe du séquençage de Sanger (Michel G Gauthier 2007)
Séquençage de nouvelle génération
Séquençage à haut débit (HTS), également connu sous le nom de Séquençage de nouvelle génération La séquençage de nouvelle génération (NGS), ou séquençage de deuxième génération, intègre principalement la technique 454 de Roche, la technique HiSeq d'Illumina, la technique SoLiD d'ABI et la technique Ion Torrent de Thermo Fisher. Les techniques 454 et SoLiD ont été abandonnées en raison de leur complexité, de leur coût élevé et de leur stabilité de séquençage incohérente. Les technologies NGS largement utilisées sur le marché actuel proviennent principalement d'Illumina et de Thermo Fisher ; des entreprises comme BGI promeuvent également des produits NGS.
En 2008, Applied Biosystems (ABI) et Invitrogen ont fusionné pour créer Life Technologies, qui a ensuite lancé les séquenceurs Ion Torrent et Ion Proton en 2010 et 2012, respectivement. En 2014, Life Technologies a été acquis par Thermo Fisher Scientific pour un montant substantiel de 13,6 milliards de dollars. À la suite de cette transaction, Thermo Fisher a massivement fabriqué et vigoureusement promu les produits phares de Life Technologies, en particulier leurs séquenceurs de qualité clinique.
En attendant, en 2014, Life Technologies a été acquis par la Thermo Fisher Corporation pour une somme substantielle de 13,6 milliards de dollars. Cette acquisition significative a transformé la fonction des principaux produits de Life Technologies. Initialement utilisés à des fins de recherche, ces dispositifs de séquençage ont été transformés en séquenceurs de qualité clinique, produits en masse et intégrés de manière transparente dans le répertoire de Thermo Fisher. La décision prise par Thermo Fisher de promouvoir ces produits était un témoignage de leur potentiel à transformer le paysage de la génomique clinique.
L'avènement de la technologie de séquençage 454 a permis d'atténuer les lacunes inhérentes aux approches de séquençage de première génération. Elle présente des caractéristiques distinctives telles qu'un débit élevé (avec une production moyenne atteignant jusqu'à 0,5 G), une rentabilité et des processus économes en temps. Cependant, lors du séquençage de séquences riches en structures nucléotidiques répétitives ou de séquences, le Séquençage de Sanger la méthodologie s'avère plus appropriée.
La technologie de séquençage Solexa a gagné en notoriété pendant une période caractérisée par des avancées rapides dans le domaine des puces génétiques et a depuis trouvé de nombreuses applications dans le séquençage des animaux, des plantes et des microbes. Semblable à la technologie de séquençage 454, Solexa fonctionne sur le principe du "séquençage par synthèse", offrant des avantages tels que l'élimination du besoin de synthèse de sondes et d'une compréhension de la séquence du génome de l'organisme modèle, simplifiant ainsi les procédures.
De plus, cette technologie utilise des puces intégrées lors de la phase de préparation pour connecter des molécules d'ADN avec des amorces spécifiques, facilitant leur liaison spécifique à la puce. Cela entraîne la production d'ADN "monoclonal" et confère des caractéristiques telles qu'un haut débit (avec une sortie moyenne atteignant jusqu'à 30G), un niveau de sensibilité élevé et une précision améliorée de la technique.
La méthode de séquençage SOLiD (Supported Oligo Ligation Detection) représente une approche technologique divergente par rapport aux stratégies précédemment mentionnées. Son principe sous-jacent repose sur la ligation plutôt que sur l'amplification par PCR. La séquence d'ADN ciblée est obtenue par plusieurs cycles de séquençage après la liaison de l'ADN cible avec des sondes fluorescentes à octa-base. En éliminant l'étape de "synthèse", cette méthode minimise efficacement les phénomènes de désaccord lors de l'appariement des bases, posant ainsi les bases d'une précision améliorée.
En comparaison avec Séquençage de Sanger, Séquençage de nouvelle génération La séquençage de nouvelle génération (NGS) a considérablement progressé en termes de débit, surmontant le modus operandi de la génération précédente qui limitait le séquençage à un seul morceau d'ADN à la fois. Un seul passage de NGS peut simultanément révéler des centaines de milliers à des millions de séquences de nucléotides, réduisant ainsi de manière spectaculaire le temps nécessaire au séquençage à l'échelle du génome. De plus, l'application généralisée de la technologie NGS est largement attribuable à son coût réduit.
Service qui pourrait vous intéresser
Séquençage à lecture longue
Le Séquençage à lecture longue des techniques, connues sous le nom de technologie de temps réel à molécule unique (SMRT) de PacBio et de technologie Nanopore d'Oxford Nanopore Technologies, répondent aux lacunes associées à Séquençage de nouvelle génération (NGS). Malgré la révolution en termes de débit offerte par le NGS, ses limitations inhérentes concernant la courte longueur des lectures créent des obstacles lors du séquençage et de l'assemblage des régions de séquences hautement répétitives. De plus, obtenir des informations précises sur la séquence des gènes à partir du NGS nécessite non seulement une couverture de séquençage élevée, mais aussi des techniques d'assemblage de séquences précises.
Séquençage à lecture longue les technologies offrent une solution à ces limitations, fournissant des résultats de séquençage pour des séquences longues individuelles de novo. Ces technologies, également connues sous le nom de séquençage de molécules uniques techniques, accomplir cet exploit en réalisant le séquençage sur de longues séquences uniques, contournant ainsi la nécessité d'une amplification PCR de la molécule cible. Le résultat est que ces méthodes fournissent directement des informations de séquence d'acide nucléique s'étendant sur des dizaines de milliers de paires de bases tout en garantissant un haut débit.
En octobre 2015, Pacific BioSciences (PacBio) a introduit le système Sequel, un séquenceur de troisième génération développé autour de leur technologie Single Molecule Real-Time (SMRT). Le Sequel se caractérise par une puce en métal dotée de nombreux guides d'ondes en mode zéro (ZMW) - en réalité des ouvertures circulaires d'un diamètre de seulement 100 nm - contenant l'ADN polymérase, l'échantillon d'ADN à séquencer et des triphosphates de désoxyribonucléotide (dNTP) marqués par fluorescence. Les canaux ZMW concentrent la lumière pertinente de manière intense dans une minuscule zone de détection au fond, capable d'enregistrer la fluorescence distincte de chaque type de dNTP, déterminant ainsi la séquence de bases de l'acide nucléique.
L'efficacité de la technologie SMRT repose sur trois composants clés : 1) La polymérase d'ADN, dont la longueur de la lecture séquencée dépend principalement de l'activité enzymatique de la polymérase. Étant donné qu'elle est sensible aux dommages causés par le laser utilisé pour la détection, c'est une considération majeure. 2) Le marquage des dNTPs avec des entités fluorescentes à l'extrémité phosphate 3'. À mesure que la synthèse de l'ADN progresse, la rupture de la liaison phosphodiester 3' libère les entités marquées utilisées pour la détection, minimisant ainsi l'encombrement stérique et favorisant une synthèse d'ADN ininterrompue. Cela garantit l'extension de la longueur de la lecture. 3) La fonction de distinction des ouvertures du ZMW, qui s'avèrent utiles pour discerner les signaux de réaction au sein du bruit de la forte fluorescence environnante. Sa solidité structurelle est soulignée par la plage d'énergie limitée due à la lumière laser qui ne peut pas pénétrer les minuscules orifices pour atteindre la zone supérieure de la solution. Cela garantit que les signaux fluorescents proviennent uniquement de la petite zone de réaction, laissant les monomères à l'extérieur du pore dans l'obscurité, minimisant ainsi efficacement la fluorescence de fond.
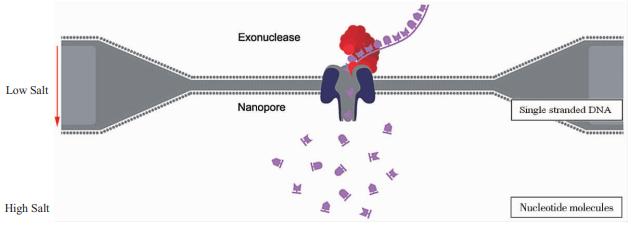 Technologie de séquençage par nanopore (Source de l'image : Nanopore)
Technologie de séquençage par nanopore (Source de l'image : Nanopore)
Simultanément, Oxford Nanopore Technologies a lancé son séquenceur de troisième génération, le MinION, reposant sur la technologie des nanopores. Les modèles opérationnels incluent une bicouche lipidique spéciale avec des électrodes d'un côté, posée au-dessus d'un nanopore. Cette bicouche contient divers nanopores constitués de protéines alpha-hémolysine, chacun accueillant une exonucléase d'acide nucléique. Le modèle d'ADN se déplaçant dans le nanopore se lie à l'exonucléase, qui coordonne pour couper successivement les bases d'ADN transitant à travers le nanopore. Lorsque chaque base passe, elle engendre un blocage de courant unique. Collectivement, la détection et l'interprétation de ces minuscules variations de courant permettent de déterminer le type de base associé, aboutissant à l'élucidation de la séquence de l'ensemble de la molécule d'ADN.
Tableau 1 Comparaison des avantages et des inconvénients du séquençage de nouvelle génération et du séquençage à longues lectures
| Séquençage de nouvelle génération | Séquençage à lecture longue | |
| Avantages | - Capable de générer plusieurs lectures pour la même région avec une profondeur de séquençage élevée, atteignant une grande précision. | - Des lectures plus longues sans besoin de références de modèles, réduisant les coûts d'assemblage en bioinformatique, économisant de la mémoire et du temps de calcul. Peut être utilisé pour déterminer des séquences hautement répétitives, améliorant la couverture de séquençage. |
| - Procédures de traitement d'échantillons matures avec un traitement d'échantillons d'ADN et des réactifs standardisés. | - Séquences directement à partir de l'échantillon d'ADN original, évitant les erreurs introduites par l'amplification PCR. | |
| - Les coûts de séquençage ont considérablement diminué, variant de 0,05 à 0,15 $ par Mb. | - Application élargie de la technologie de séquençage, par exemple, le séquençage direct des séquences d'ARN, réduisant considérablement les erreurs système introduites par la transcription inverse. Séquençage direct des séquences d'ADN méthylé, distinguant entre les bases C normales et méthylées en mesurant les temps de pause de l'ADN polymérase. | |
| - Logiciel de bioinformatique hautement mature pour un traitement rapide des données, le contrôle de la qualité et l'assemblage. | - Applicable au ctDNA et au séquençage à cellule unique : La haute sensibilité du séquençage de 3ème génération permet de surveiller le ctDNA à des niveaux inférieurs à 1 ng. Au niveau de la cellule unique, le séquençage direct de l'ADN original permet un séquençage in situ après lyse cellulaire. | |
| Inconvénients | - Longueurs de lecture plus courtes, assemblage chronophage et couverture plus faible des régions de séquences répétitives et palindromiques. | - Des taux d'erreur plus élevés dans les longueurs de lecture unique, nécessitant un séquençage répété pour la correction des erreurs, augmentant considérablement les coûts de séquençage. |
| - Introduction d'erreurs systématiques lors de la synthèse et du séquençage utilisant la technologie PCR. | - Forte dépendance à l'activité des enzymes biologiques, les conditions de séquençage affectant directement l'efficacité et la précision du séquençage. | |
| - Dépendance à des processus en amont tels que la transcription inverse et le traitement au bisulfite pour le séquençage de génomes non-ADN (par exemple, le séquençage de l'ARN, le séquençage de la méthylation de l'ADN), ce qui introduit un risque accru d'erreurs. | - Coût plus élevé, variant de 0,33 $ à 1,00 $ par 1 Mo. | |
| - Sensibilité réduite pour les échantillons d'ADN à faible abondance et assemblage difficile sans modèles de référence préexistants. | - Variété insuffisante dans les logiciels d'analyse bioinformatique et accumulation de données limitée. |
Ces dernières années ont été marquées par une adoption robuste de Séquençage à long terme technologies dans les analyses génomiques. Ces méthodologies ont considérablement propulsé le progrès de l'exploration et du déchiffrement génomiques, réalisant des avancées substantielles. Le séquençage à longues lectures, le séquençage direct de molécules d'ADN individuelles, offre des longueurs de lecture plus importantes et permet une sensibilité élevée, un assemblage sans modèle et le séquençage direct des méthylomes d'ADN et de l'ARN. Cependant, des limitations existent. Des contrôles opérationnels précis sont nécessaires, les coûts sont substantiels, les logiciels de traitement bioinformatique matures sont rares et les taux d'erreur sont élevés. Surtout dans des contextes exigeant des coûts et une précision élevés, tels que la médecine de précision et le dépistage précoce du cancer, l'application pratique de ces technologies reste contrainte.
Au contraire, tandis que Séquençage de nouvelle génération (NGS) présente des inconvénients, notamment la dépendance à des modèles existants, la courte longueur des lectures et le biais de séquence, mais son système mature, sa capacité de haut débit, sa haute sensibilité, sa haute résolution et son faible coût le rendent plus accessible et largement adopté dans divers laboratoires. En effet, la relation entre Séquençage à lecture longue et le NGS n'est pas caractérisé par la concurrence ou le remplacement, mais plutôt, ces technologies agissent comme des actifs complémentaires.
En prenant l'exemple du séquençage des groupes de résistance microbienne, la plupart des gènes de résistance se trouvent sur des plasmides, dont beaucoup ne sont pas correctement séquencés lors des recherches préliminaires. Une dépendance exclusive au séquençage de nouvelle génération (NGS) ne peut pas obtenir avec précision les séquences de plasmides contenant des gènes de résistance. À ce stade, le séquençage à longues lectures peut être utilisé pour séquencer directement l'ADN à molécule unique du plasmide. Cependant, en raison de la faible précision et exactitude du séquençage à longues lectures, une fois la séquence du plasmide obtenue, il est souvent nécessaire d'utiliser le NGS pour générer un grand nombre de courtes lectures de haute précision pour comparaison, ce qui conduit à une séquence de plasmide plus précise.
Alors que le coût du séquençage à longues lectures continue de diminuer, de plus en plus de scénarios nécessiteront la combinaison des technologies NGS et de séquençage à longues lectures pour obtenir des séquences inconnues précises dans les futures applications cliniques. Ces deux technologies peuvent se compléter, renforçant mutuellement leurs valeurs d'application respectives.
Service qui pourrait vous intéresser
Principes et processus de la technologie NGS
En 2005, le système Genome Sequencer 20 (GS 20) a été introduit par la société 454, ouvrant l'ère du "Séquençage par synthèse" (SBS). Aujourd'hui, la méthodologie SBS est fondamentale pour les plateformes de séquençage de nouvelle génération (NGS) Ion Torrent de Thermo Fisher et Illumina.
L'essence de la technologie SBS est commune à toutes les plateformes et se compose principalement de trois étapes clés : 1) La préparation de la bibliothèque, qui consiste à fragmenter des échantillons d'ADN à longues chaînes en segments courts pour créer une bibliothèque d'ADN. Lors de la fragmentation, des adaptateurs sont fixés aux deux extrémités des segments courts pour faciliter la fixation ultérieure des fragments, en utilisant les adaptateurs comme sites de liaison pour les amorces lors des processus d'amplification ; 2) Le chargement, dans lequel la bibliothèque d'ADN est répartie sur un support de séquençage et subit une amplification. L'objectif de cette étape est de multiplier le nombre de ces segments courts, générant une quantité suffisante de fragments courts identiques pour amplifier les signaux lors de la phase de séquençage suivante. Cela permet la détection des informations de séquence ; 3) Le séquençage, où l'ADN double brin amplifié subit une dénaturation pour former de l'ADN simple brin, qui sert ensuite de modèle. L'intégration des nucléotides simples lors de chaque cycle de synthèse est surveillée et les informations de séquence sont discernées à partir de l'enregistrement des signaux (par exemple, fluorescence, variations de pH).
Les différences entre les produits NGS SBS proposés par différentes entreprises se manifestent dans des aspects tels que le traitement des échantillons, la détection des signaux et la fixation des fragments.
Les procédures standard pour Séquençage de nouvelle génération les technologies comprennent les suivantes :
Pour la technologie de séquençage 1.3.1454, le processus commence par la fragmentation de l'échantillon, suivie de la sélection de fragments d'une longueur comprise entre 500 et 800 paires de bases (pb). Des adaptateurs sont ensuite ajoutés aux deux extrémités de ces fragments. Les fragments d'ADN ligaturés avec des adaptateurs sont combinés avec des billes magnétiques, encapsulés dans un système eau-dans-huile, et associés à une plaque de réaction. Le système est ensuite intégré dans une plaque PTP où les quatre types de bases sont ajoutés séquentiellement pour le séquençage. Enfin, les signaux sont surveillés et les données de séquence collectées.
La technologie de séquençage Solexa commence également par la fragmentation de l'échantillon. Par la suite, une bibliothèque est établie et amplifiée sur une puce intégrée avec des amorces pour produire des clones uniques. Le système introduit ensuite quatre dNTP étiquetés différemment ainsi qu'une ADN polymérase modifiée. Les signaux fluorescents collectés sont enregistrés pour extraire les données de séquence.
La technologie de séquençage Solid commence par la fragmentation de l'échantillon et l'attachement d'adaptateurs aux deux extrémités de ces fragments. Cela est suivi par une amplification PCR et un enrichissement par microparticules. Des billes magnétiques sont ensuite ajoutées, permettant l'hybridation entre les adaptateurs et des séquences d'adaptateurs spécifiques sur le modèle. Quatre sondes à double base teintées fluorescentes de couleurs différentes sont connectées à l'échantillon. L'étape finale consiste à détecter les signaux fluorescents et à recueillir les données de séquence.
En s'appuyant sur les descriptions ci-dessus, nous avons juxtaposé les caractéristiques de trois technologies de séquençage de deuxième génération. Séquençage de Sanger technologie (tableau 1). Une comparaison horizontale des technologies de séquençage de deuxième génération a également été réalisée (tableau 2). On peut en déduire qu'en comparaison avec le séquençage de première génération Sanger, toutes les technologies de séquençage de deuxième génération montrent un haut débit, une grande précision et une rentabilité.
Tableau 2 : Comparaison des technologies de séquençage
| Technologie | Principe | Approche de séquençage | Longueur de lecture | Précision | Types d'erreurs | Coût ($/Mo) | Débit | Plateforme |
| 454 | Pyroséquençage | Synthèse et séquençage simultanés | 500 pb | 99 % | Insertions, suppressions | Bas | 250-400 Mo | Perle |
| Solexa | Fluorescence | Synthèse et séquençage simultanés | 100 points de base | 98,99 % | Substitutions | Bas | 3-6 Go | Verre |
| Solide | Fluorescence | Séquençage basé sur la ligature | 50 points de base | 99,94 % | Substitutions | Très bas | 30 Go | Verre |
| Sanger | Radioactif | Synthèse et séquençage simultanés | 1000 bp | 99,99 % | Substitutions | Élevé | 1 Mo | PAGE |
Tableau 3 : Comparaison de trois technologies de séquençage de nouvelle génération
| Technologie de séquençage | Avantages | Inconvénients |
| Roche/454 | Longueurs de lecture élevées | Faible débit, coût élevé |
| Solexa | Haute capacité, haute précision, faible coût | Longueurs de lecture courtes, opération complexe |
| Solide | Haute capacité, haute précision, faible coût | Longueurs de lecture courtes, opération complexe |
Contrôle de la qualité des données pour le séquençage de nouvelle génération (NGS)
Le processus de séquençage dans la synthèse par séquençage (SBS) présente un problème notable : à mesure que la chaîne synthétisée s'allonge, l'efficacité de l'ADN polymérase diminue, réduisant simultanément sa spécificité. Cela entraîne une augmentation progressive du taux d'erreurs de synthèse des bases à mesure que le séquençage progresse, soulignant l'importance du contrôle de qualité (CQ) dans Séquençage de nouvelle génération données (NGS) pour des analyses ultérieures. Cela devient également un identifiant crucial lors de l'évaluation de la cohérence des données et de la qualité des instruments de séquençage.
Dans le cadre du QC pour les résultats de NGS, les paramètres d'une importance capitale incluent :
1) Q20, Q30 et Q40 : Dans le séquençage à haut débit des gènes, chaque base se voit attribuer une valeur de qualité (Q), plus la valeur Q est élevée, moins il y a de chances qu'une mesure de base soit erronée, calculée par la formule Q = -10logP. Q20 et Q30 représentent la proportion de bases avec des valeurs de qualité égales ou supérieures à 20 ou 30, respectivement.
2) Profondeur de séquençage : Se réfère au rapport entre le nombre total de bases obtenues par séquençage et la taille du génome cible. Si la taille du gène est de 2M et que la profondeur de séquençage est de 10X, la quantité totale de données obtenues s'élève à 20M. Ainsi, la profondeur de séquençage est calculée comme Données Totales (20M) / Taille du génome (2M) = 10X.
3) Couverture de séquençage : Elle définit la proportion de séquences obtenues par séquençage par rapport à l'ensemble du génome. Étant donné la présence de structures complexes, comme des sections avec un contenu en GC élevé et des séquences répétées dans le génome, les séquences assemblées après séquençage ne couvriraient pas toutes les zones. Les régions dépourvues de séquences sont appelées "lacunes". Par exemple, si la couverture de séquençage d'un génome bactérien est de 98 %, 2 % de la région de séquence reste non séquencée.
Technologies dérivées pour le séquençage de nouvelle génération (NGS)
Bien que Séquençage de nouvelle génération (NGS) à lui seul ne produit que des séquences d'ADN, mais il peut générer un éventail d'informations, y compris des transcriptomes ciblés, des méthylomes et des métagénomes lorsqu'il est combiné avec diverses techniques de traitement d'échantillons (telles que la transcription in vitro inversée, la capture de séquences et le traitement au bisulfite). La corrélation de ces techniques est encore facilitée par des analyses bioinformatiques ultérieures. Avec la précision à haut débit offerte par le NGS, un vaste éventail de perspectives cliniques est envisageable. Il présente des applications potentielles dans divers scénarios cliniques tels que le dépistage du cancer, les tests génétiques, la médecine de précision et l'amélioration des cultures, élargissant ainsi considérablement l'étendue de son utilité (voir Tableau 4).
Tableau 4. Technologies dérivées du séquençage NGS et leurs applications cliniques
| Flux de travail | Objectif de séquençage | Scénarios d'application |
| RNA-Seq | Transcription inverse de l'extrait du transcriptome, suivie du séquençage NGS de l'ADNc résultant, pour obtenir des profils d'expression génique au sein des cellules cibles. | Dépistage précoce du cancer, diagnostic précoce des maladies métaboliques, diagnostics compagnons, etc. |
| ChIP-Seq | Utilise l'immunoprécipitation de la chromatine (ChIP) pour isoler les fragments d'ADN liés à la protéine cible. Après élution, le séquençage NGS révèle la séquence des sites de liaison pour la protéine cible. | Identification des sites de liaison pour les facteurs de transcription cibles, découverte de biomarqueurs et élucidation des réseaux de régulation génique. |
| Séquençage des exons et des régions ciblées | Utilise la technologie de capture de séquence pour capturer et enrichir l'ADN des exons de l'ensemble du génome ou d'autres régions spécifiques, suivie du séquençage de nouvelle génération (NGS). | Obtention d'informations de séquence telles que des SNPs et des variations structurelles dans des régions spécifiques à un coût relativement bas ; utilisé dans le diagnostic génétique néonatal et plus encore. |
| Séquençage du génome entier | Extrait l'ADN génomique d'une population mixte pour le séquençage NGS, obtenant les génomes core et variables des espèces mélangées. | Analyse des séquences de séquençage, y compris l'ADNr 16S, pour recueillir des informations sur la composition de la population et du phénotype de l'échantillon. Utilisé dans l'élevage des cultures, la transplantation du microbiome humain, etc. |
| Séquençage de méthylation | Applique un traitement au bisulfite de sodium à l'ADN, convertissant la cytosine (C) non méthylée en uracile (U). Un séquençage NGS du génome entier est ensuite effectué. | Atteint une résolution à base unique au niveau du génome entier, évaluant avec précision les niveaux de méthylation des bases C individuelles et construisant des cartes détaillées de méthylation de l'ADN. Utilisé dans le dépistage précoce du cancer, le diagnostic précoce des maladies métaboliques, les diagnostics compagnons, la pharmacogénomique, etc. |
| Études d'association à l'échelle du génome | Basé sur l'analyse des SNV obtenue par séquençage du génome entier, évalue sa corrélation avec des traits spécifiques (maladies, réponses immunitaires) dans des populations spécifiques. | Identification des SNV qui déterminent des traits difficiles à obtenir par des techniques moléculaires traditionnelles. Utilisé pour estimer les risques de maladies génétiques, la détection des tumeurs, le dépistage précoce du cancer, la médecine de précision, etc. |
Références :
- Maxam A M, Gilbert W. Une nouvelle méthode pour séquencer l'ADN. Proc Natl Acad Sci États-Unis, 1977, 74(2) : 560-564.
- Hillier L W, Marth G T. Quinlan A R, et al. Séquençage du génome entier et découverte de variants chez C. elegans. Méthodes Nat, 2008, 5 : 183-188.
- D Y Zhang, T X Zhang, G X Wang. Développement et application de la technologie de séquençage de deuxième génération. Sciences et technologies de l'environnement, 2016, 39(9) : 96-102.
- H CHEN, X F TAN. Excavation des ressources génétiques basé sur le séquençage de nouvelle génération. Journal de Physiologie Végétale des Technologies, l 2014, 50(8) : 1089-1095.
- Pacific Biosciences of California Inc. USA sur le web URL [EB/OL]. 2017, 20 mai. http://www.pacb.oom/science/smrt-sequencing/.
- Clarke J, Wu H C, Jayasinghe L, et al. Identification continue de bases pour le séquençage de l'ADN par nanopore à molécule unique. Nature Nanotechnology, 2009, 4(4) : 265-270.
- P LU, J J JIN, Z F LI, P J CAO, et al. Assemblage du génome basé sur la technologie de séquençage à longues lectures et son application dans le tabac. Science et Technologie du Tabac, 2018, 02 : 0087-0108.
- Haas B J, Zody M C. Avancer l'analyse RNA-Seq. Nat Biotechnol2010, 28(5) : 421-423.
- Cock Peter J A, Fields Christopher J, Goto Naohisa Le format de fichier FASTQ de Sanger pour les séquences avec des scores de qualité, et les variantes FASTQ de Solexa/Illumina. Recherche sur les acides nucléiques, 2009, 38(6) : 1767-1771.
- L K Wang, Procession et application des données RNA-Seq [D]. 2012, 6.
- Z P Wu, X Wang, X G Zhang. Utilisation de modèles de distribution de lecture non uniforme pour améliorer l'inférence de l'expression des isoformes dans le RNA-Seq. Bioinformatique (En ligne), 2010, 27(4) : 502-508.
- Hansen Kasper D, Brenner Steven E, Dudoit Sandrine Biais dans le séquençage de transcriptome Illumina causés par le priming aléatoire avec des hexamères. Recherche sur les acides nucléiques, 2010, 38(12), e131.
- He M, Y Wang, W P Hua, Y Z Zhang, Z Z Wang. Séquençage de novo du transcriptome de Hypericum perforatum pour identifier des gènes potentiels impliqués dans la biosynthèse de métabolites actifs. PLoS ONE, 2012, 7(7) : e42081.
- M Z Wang, B Wu, C H Chao et S F Lu. Identification des ARN non codants de type mARN et validation d'un puissant nommé MAR dans le Panax ginseng. Journal de la biologie intégrative des plantes, 2015, 57(3) : 256-270


