
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Traçage de la lignée mitochondriale dans les modèles RUO : des séquences ancestrales à l'authentification des lignées cellulaires
Les modèles de recherche ne sont fiables que dans la mesure où leurs dossiers d'identité le sont. En pratique, cette confiance peut s'éroder progressivement plutôt que d'échouer d'un seul coup : une lignée cellulaire peut rester morphologiquement familière tandis que son profil d'hétéroplasmie mitochondriale évolue au fil des passages successifs, ou une population contaminante à faible niveau peut rester inaperçue jusqu'à ce qu'une expérience en aval devienne difficile à reproduire. Pour la gouvernance des modèles RUO, le séquençage de l'ADN mitochondrial (ADNmt) ajoute une couche de preuve utile car l'ADNmt est hérité de manière maternelle, présent en de nombreuses copies par cellule, et bien adapté à un examen de lignée au niveau du séquençage lorsque les équipes ont besoin de plus qu'un simple instantané d'identité. La littérature récente axée sur les RUO montre que les variantes d'ADNmt peuvent fonctionner comme des marqueurs de lignée endogène dans des conditions analytiques définies, bien que l'interprétation dépende de l'expansion clonale, de la dynamique d'hétéroplasmie et des seuils validés.
Cet article explique comment le traçage de lignée de l'ADNmt s'intègre dans l'authentification du modèle RUO, ce que signifie l'expression. "la séquence d'ADN mitochondrial ancestrale représente théoriquement" signifie dans le travail phylogénétique pratique, comment un base de données de séquences d'ADN mitochondrial soutient l'attribution de haplogroupe et la révision de contamination, et comment évaluer un service de séquençage de l'ADN mitochondrial lorsque le véritable objectif est la gestion de modèles reproductibles plutôt que le séquençage ponctuel.
La base biologique : Pourquoi l'ADNmt est un "code-barres moléculaire" pour les modèles RUO
L'ADN mitochondrial est souvent décrit comme un code-barres moléculaire car il combine trois propriétés particulièrement utiles dans les workflows RUO : l'hérédité matrilinéaire, l'accumulation relativement rapide de variations et un nombre élevé de copies par cellule. MITOMAP décrit le génome mitochondrial humain comme une molécule circulaire de 16 569 nucléotides et continue de servir de principal hub de référence pour les outils et la curation de l'ADNmt. En termes pratiques, cela signifie qu'un profil d'ADNmt bien caractérisé en début de passage peut servir de référence de lignée contre laquelle les échantillons ultérieurs sont vérifiés pour la continuité, la divergence ou les signaux de lignées mixtes.
Cela est particulièrement utile lorsque la question de contrôle qualité va au-delà de la confirmation des espèces pour inclure la continuité des lignées par rapport à une référence interne établie pour le même modèle de recherche. Dans un projet qui nécessite un contexte génomique plus large en parallèle avec l'examen mitochondrial, Séquençage du génome entier peut fournir un contexte à l'échelle du génome, tandis que Séquençage de l'ADN mitochondrial (ADNmt) est le meilleur ajustement direct lorsque l'objectif immédiat est le profilage mitochondrial sensible à la lignée.
Un deuxième concept qui mérite une explication attentive est la phrase "la séquence d'ADN mitochondrial ancestrale représente théoriquement." En termes phylogénétiques, cela fait référence à un état de référence ancestral inféré près de la racine de l'arbre mitochondrial humain moderne, et non à un échantillon moderne observé. C'est la logique derrière le Séquence de référence sapiens reconstruite (RSRS)tandis que le séquence de référence de Cambridge révisée (rCRS) reste le cadre de coordonnées historiquement adopté utilisé dans de nombreux pipelines de reporting. Pour les projets RUO, le point clé est opérationnel : le choix de référence affecte la manière dont les variantes sont écrites, mais l'attribution de l'haplogroupe dépend du contexte phylogénétique, et non d'un simple comptage de différences à partir d'un seul standard de coordonnées.
Cette distinction est importante lors de l'examen d'un rapport de fournisseur. Un rapport utile devrait indiquer quelle séquence de référence a été utilisée pour l'alignement et la notation, quelle base de données ou cadre phylogénétique a soutenu la classification des haplogroupes, et si l'interprétation était basée sur l'ADNmt complet, des régions ciblées ou uniquement des loci de points chauds.
Comparé à l'authentification basée sur les STR de routine, le séquençage de l'ADNmt n'est pas la norme universelle pour les tests d'identité. L'ICLAC continue de positionner le profilage STR comme une norme fondamentale pour de nombreux workflows d'authentification des lignées cellulaires humaines. Cependant, l'ADNmt peut apporter une valeur importante lorsque la question concerne la continuité de la lignée maternelle, la récupération à faible apport, l'examen de la contamination sensible à la lignée ou le suivi conscient de l'hétéroplasmie au fil du temps.
Atténuer les risques dans la recherche longitudinale : dérive génétique et contamination croisée
 Figure 1. Interprétation de la dérive de l'ADNmt par rapport à la contamination croisée lors du passage en série dans des modèles RUO. La figure contraste le changement progressif d'hétéroplasmie au sein de l'arrière-plan de lignage attendu avec l'apparition de combinaisons de marqueurs inattendues plus cohérentes avec une contamination par lignages mixtes.
Figure 1. Interprétation de la dérive de l'ADNmt par rapport à la contamination croisée lors du passage en série dans des modèles RUO. La figure contraste le changement progressif d'hétéroplasmie au sein de l'arrière-plan de lignage attendu avec l'apparition de combinaisons de marqueurs inattendues plus cohérentes avec une contamination par lignages mixtes.
L'utilisation longitudinale crée deux risques liés mais distincts : dérive génétique et contamination croiséeLa dérive apparaît généralement comme des variations des fréquences d'hétéroplasmie au fil du temps. Un variant présent à faible fréquence dans un échantillon conservé tôt peut s'étendre, se contracter ou disparaître dans des passages ultérieurs, car les génomes mitochondriaux se répartissent de manière stochastique et parce que différents sous-clones ne contribuent pas de manière égale aux passages futurs. Des travaux récents dans Biologie des génomes met l'accent sur le fait que de nombreux variants mtDNA informatifs sur la lignée sont des hétéroplasies préexistantes plutôt que des mutations nouvellement générées, et que l'utilité de ces marqueurs dépend fortement de l'expansion clonale.
La contamination croisée est différente. Ici, le signe d'alerte est l'apparition de combinaisons de marqueurs qui ne correspondent pas à l'arrière-plan de lignée attendu, comme un schéma de haplogroupe inattendu ou un profil mixte qui ne peut pas être expliqué uniquement par une hétéroplasmie précédemment documentée. En termes opérationnels de RUO, cela signifie que les équipes doivent définir une ligne de base d'ADNmt à l'entrée du modèle, archiver les données brutes et comparer les échantillons ultérieurs par rapport à la ligne de base plutôt qu'à un dossier de laboratoire simplifié.
Pour l'optimisation des protocoles dans des conditions de faible entrée ou de matrices lourdes, voir Protocole de séquençage de l'ADN mitochondrial pour des échantillons complexesCela est particulièrement pertinent lorsque le passage en série produit un matériel stressé, à faible rendement ou d'une composition inégale.
D'un point de vue de planification des services, le flux de travail le plus efficace est généralement sélectif plutôt qu'exhaustif. La confirmation d'identité en amont peut se connecter à Identification de lignées cellulairesla récupération mitochondriale ciblée peut s'adapter Séquençage de région cibléeet un suivi quantitatif de l'abondance mitochondriale peut se connecter à Test de quantification du nombre de copies d'ADN mitochondrial.
Quand utiliser les vérifications de lignée mtDNA
Utilisez des vérifications de lignée d'ADNmt lorsque le modèle a subi un passage sériel prolongé, lorsque le matériel est d'entrée faible ou partiellement compromis, lorsque la divergence phénotypique inexpliquée soulève des inquiétudes quant à la cohérence du modèle, ou lorsqu'une référence mitochondriale au niveau de la séquence est nécessaire pour des comparaisons futures. Dans ces contextes, l'ADNmt ajoute un contexte sensible à la lignée que les contrôles d'identité de routine peuvent ne pas capturer à eux seuls.
Quand ne pas surinterpréter l'ADNmt seul
Ne considérez pas l'ADNmt seul comme une réponse complète lorsque la tâche immédiate consiste à faire correspondre l'identité des lignées cellulaires humaines de routine, lorsque les seuils d'hétéroplasmie ne sont pas validés pour la plateforme et la profondeur utilisées, lorsque le rapport manque de références claires ou de versionnage de base de données, ou lorsque des preuves du génome nucléaire sont nécessaires pour résoudre l'ambiguïté. L'ADNmt est le plus utile lorsqu'il est présenté comme un élément d'une logique de gouvernance de modèle plus large plutôt que comme une solution universelle autonome.
Avant de commander une course de séquençage, confirmez quatre points de décision : si un échantillon de référence du même modèle est disponible, si la plage d'hétéroplasmie attendue est compatible avec la plateforme et la profondeur, si un examen de lignage mixte est nécessaire, et si la question peut être résolue uniquement par l'ADNmt. Ce court pré-contrôle aide à prévenir qu'une course techniquement réussie ne produise un rapport opérationnellement faible.
| Question préalable | Pourquoi c'est important | Impact sur le choix de la méthode |
|---|---|---|
| Un échantillon de référence du même modèle est-il disponible ? | La comparaison de lignées directes est beaucoup plus solide que l'interprétation autonome. | Soutient l'examen de la continuité de l'ADNmt |
| Les hétéroplasmiques à faible fréquence ont-elles de l'importance ? | La révision des variantes mineures dépend de la profondeur et des seuils validés. | Peut nécessiter un séquençage plus approfondi ou un contrôle qualité plus strict. |
| Une révision de lignée mixte est-elle requise ? | L'examen de la contamination nécessite une interprétation explicite, pas seulement un appel au consensus. | Nécessite un reporting prenant en compte la contamination |
| Le contexte nucléaire est-il également nécessaire ? | Certaines questions dépassent le champ mitochondrial. | Peut justifier une génomique plus large en parallèle. |
Exploitation des bases de données de séquences d'ADN mitochondrial pour l'authentification
L'authentification devient plus défendable lorsque les données de séquence brutes sont interprétées par rapport à des ressources mitochondriales soigneusement sélectionnées plutôt qu'à un appariement ad hoc de SNP. En pratique, trois couches devraient rester connectées : l'alignement de référence, la classification phylogénétique et l'interprétation consciente de la base de données.
Tout d'abord, la plupart des flux de travail décrivent encore les variantes en utilisant un cadre orienté rCRS. MITOMAP connecte explicitement les utilisateurs au cadre d'accès rCRS et fournit des outils tels que MITOMASTER pour l'interprétation des séquences d'ADNmt. Deuxièmement, l'attribution des haplogroupes repose sur la structure phylogénétique, et non sur une simple distance par rapport à une référence. PhyloTree reste une ressource fondamentale en phylogénie de l'ADNmt, et le site identifie actuellement la Build 17 datée du 18 février 2016. C'est utile, mais cela signifie également qu'un fournisseur devrait être en mesure d'expliquer si sa logique de classification est basée uniquement sur l'ancienne version ou complétée par des pratiques de curation plus récentes.
La valeur pratique d'une base de données de séquences d'ADN mitochondrial dans l'authentification RUO ne réside pas uniquement dans la taxonomie. Elle aide à répondre à trois questions : l'échantillon se classe-t-il là où la référence suggère qu'il devrait se classer, les marqueurs observés pointent-ils vers un schéma haplogroupe cohérent, et des variantes mineures ou des combinaisons de marqueurs soulèvent-elles une préoccupation de lignage mixte ?
Pour de nombreux projets, une norme de vérification minimale devrait inclure la séquence de référence utilisée, l'attribution de l'haplogroupe avec le cadre nommé, une liste des variantes définissantes et de soutien, un tableau d'hétéoplasmie avec les fractions alléliques et la profondeur de lecture, ainsi qu'une comparaison directe avec l'échantillon de référence interne du même modèle.
Lorsque le champ s'élargit au-delà de l'examen mitochondrial, il est préférable de ne lier que les services les plus pertinents pour la décision. Si la question reste axée sur le mitochondrial, Séquençage de l'ADN mitochondrial humain (ADNmt) est un ajustement logique ; si la question s'élargit à une caractérisation de modèle plus large, Séquençage de l'exome complet peut fournir un contexte supplémentaire sans changer la nécessité d'un cadre clair de contrôle qualité mitochondrial.
Choisir le bon service de séquençage de l'ADN mitochondrial pour le traçage de lignée
Un service de séquençage de l'ADN mitochondrial doit être évalué comme un système analytique complet, et non comme une course de séquençage isolée. La première question est profondeurL'interprétation de l'hétéroplasmie dépend de la couverture, du comportement de la plateforme et des paramètres de l'appelant. A 2024 Rapports scientifiques Une étude évaluant le séquençage de l'ADN mitochondrial par nanopore à longues lectures a rapporté que, dans cette configuration spécifique, la détection fiable de l'hétéroplasmie était d'environ 12 % à une profondeur de 150×, tout en soulignant également les différences de détection à faible fréquence dépendantes de la plateforme. Ce n'est pas un seuil universel, mais cela rappelle que les affirmations des fournisseurs doivent être liées à des conditions analytiques validées plutôt qu'à un langage de sensibilité générique.
La deuxième question est rapportabilitéUn service utile ne devrait pas se limiter à la livraison de fichiers FASTQ ou BAM. Pour le traçage de lignées et l'authentification, la sortie minimale pratique est constituée de données brutes et traitées, d'un résumé de couverture à travers le génome mitochondrial, d'un tableau de variants avec fractions alléliques et profondeur, de seuils d'interprétation de l'hétéoplasmie, d'un cadre de référence nommé, d'une attribution de haplogroupe, d'une évaluation de contamination ou de lignées mélangées, et d'une conclusion concise sur la cohérence de la lignée par rapport à la base de référence soumise.
La troisième question est transparence de la chaîne d'approvisionnementDemandez si le flux de travail traite de l'ADNmt complet par rapport à un design ciblé, de la gestion de faible complexité, de la gestion des doublons lorsque cela est pertinent, des définitions de seuils pour l'hétéoplasmie à faible fréquence et de la reproductibilité entre les réplicats techniques lorsque cela est nécessaire. Pour la confirmation orthogonale d'un petit nombre de loci prioritaires, Séquençage de Sanger peut être utile ; lorsque le projet s'étend à un contexte d'identité ou de variante plus large, Appel de variantes fournit un cadre en aval plus conscient du génome.
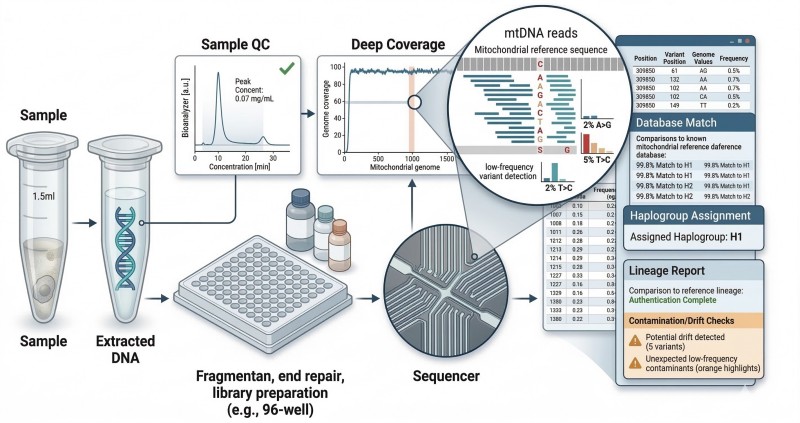 Figure 2. Flux de travail de séquençage mtDNA de bout en bout pour le traçage de lignées RUO, de l'assurance qualité des échantillons et du séquençage à l'interprétation consciente de la base de données et au rapport final.
Figure 2. Flux de travail de séquençage mtDNA de bout en bout pour le traçage de lignées RUO, de l'assurance qualité des échantillons et du séquençage à l'interprétation consciente de la base de données et au rapport final.
Un cadre décisionnel d'authentification pratique aide à prévenir la surutilisation ou la sous-utilisation des données d'ADNmt. Si l'objectif immédiat est l'appariement d'identité des lignées cellulaires humaines, le profilage STR reste la référence acceptée dans de nombreux flux de travail. Si l'objectif est la cohérence de la lignée maternelle, l'examen de l'hétéroplasmie ou la récupération à partir de matériel à faible entrée ou partiellement compromis, le séquençage de l'ADNmt peut ajouter un contexte utile au niveau de la séquence. Si la question s'étend au-delà de la cohérence de la lignée vers une caractérisation de modèle plus large, une différenciation de sous-clones ou un contexte de variantes plus large, des méthodes génomiques plus larges peuvent être plus appropriées. Dans tous les cas, l'interprétation la plus défendable provient de la comparaison de l'échantillon actuel avec une référence interne et nécessite un rapport explicite sur la profondeur, la fraction allélique, le cadre de référence et la politique de seuil.
| Cas d'utilisation | Méthode préférée | Pourquoi | Exigence minimale de rapport |
|---|---|---|---|
| Correspondance d'identité des lignées cellulaires humaines de routine | Profilage STR | Base acceptée pour de nombreux flux de travail | Interprétation des résultats STR plus correspondance |
| Revue de la cohérence de la lignée maternelle ou de l'hétéroplasmie | séquençage de l'ADNmt | Contexte de séquence sensible à la lignée | Profondeur, fraction d'allèle, haplogroupe, comparaison de base |
| Caractérisation du modèle élargie | WES/WGS avec mtDNA si nécessaire | Contexte de variante élargie et aperçu du modèle | Interprétation de la portée des variantes plus identité/QC |
| Préoccupation de lignage mixte dans la culture longitudinale | STR plus ADNmt | Combine l'examen d'identité avec la détection de signaux sensible à la lignée. | Résultat d'identité, revue de l'ADNmt, évaluation de la contamination |
Pour l'interprétation en aval après authentification, voir Analyse comparative des séquences d'ADNmt.
QC et Dépannage : Symptômes, Causes Probables et Actions
L'échec analytique le plus courant dans l'examen des lignées d'ADNmt n'est pas seulement dû à un mauvais séquençage. C'est une mauvaise formulation de la question décisionnelle. Une analyse réussie peut encore produire un rapport peu utile si la ligne de base est manquante, si la plage d'hétéroplasmie d'intérêt est inférieure à la performance validée du test, ou si l'examen de contamination n'a jamais été spécifié avant le début du séquençage.
| Symptôme | Causes probables | Action recommandée |
|---|---|---|
| Des variants inattendus à basse fréquence n'apparaissent que dans les passages tardifs. | Dérive d'hétéroplasmie, expansion de sous-clones, seuils de détection à la limite. | Vérifiez à nouveau la distribution de profondeur, comparez à la référence, envisagez une confirmation par réplicat. |
| Les marqueurs définis pointent vers plus d'un modèle d'haplogroupe. | Contamination croisée, culture mixte, artefact d'alignement | Re-séquencer du matériel frais et demander un examen explicite des lignées mixtes. |
| Les fréquences des variantes fluctuent à travers les répétitions techniques. | Faible entrée, amplification inégale, profondeur insuffisante | Augmentez l'entrée si possible et utilisez une politique de seuil validée. |
| La conclusion finale est vague malgré un large ensemble de données. | Cadre de reporting faible, pas de comparateur de base | Exiger un rapport de traçabilité orienté vers la décision |
Lorsqu'il est soutenu par une comparaison de référence, des seuils validés et une interprétation consciente de la base de données, le séquençage de l'ADNmt peut aider à distinguer la continuité de lignée des signaux de dérive ou de lignée mixte dans la gestion des modèles RUO. L'objectif n'est pas simplement de générer plus de données de séquence, mais de générer des données de séquence qui répondent à une question de contrôle qualité définie.
Conclusion : Assurer la reproductibilité dans la recherche mitochondriale
Le traçage de lignée de l'ADNmt est précieux dans la gouvernance des modèles RUO car il ajoute une vue durable au niveau de la séquence de la continuité des lignées, ce qui devient particulièrement utile lorsque les modèles sont conservés, étendus, échangés entre équipes ou revisités après de longs intervalles expérimentaux. Utilisé avec une comparaison de base et des seuils transparents, le séquençage de l'ADNmt peut aider à séparer la variation mitochondriale attendue des signaux d'alerte qui méritent un suivi.
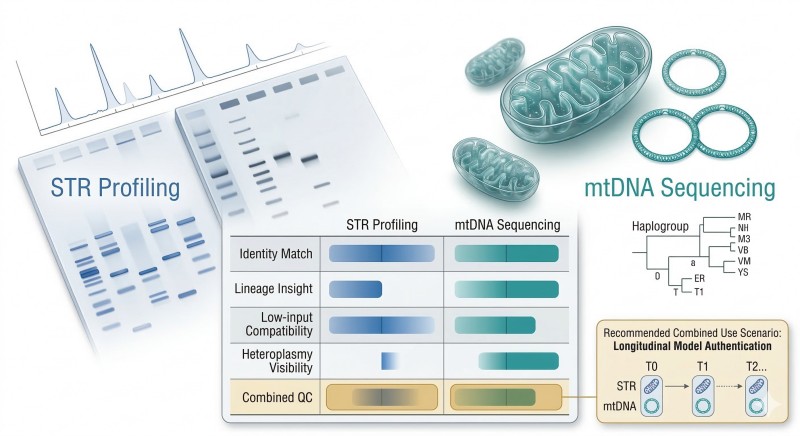 Figure 3. Rôles complémentaires du profilage STR et du séquençage de l'ADNmt dans l'authentification des modèles RUO, y compris la correspondance d'identité, le contexte de lignée, la compatibilité à faible entrée et la visibilité de l'hétéroplasmie.
Figure 3. Rôles complémentaires du profilage STR et du séquençage de l'ADNmt dans l'authentification des modèles RUO, y compris la correspondance d'identité, le contexte de lignée, la compatibilité à faible entrée et la visibilité de l'hétéroplasmie.
L'approche opérationnelle la plus robuste consiste à formaliser l'examen mitochondrial dans une procédure opérationnelle standard périodique plutôt que de le traiter comme une exception. Un modèle de mise en œuvre concis est présenté ci-dessous.
| Étape de la procédure opérationnelle standard | Action recommandée | But de la mission |
|---|---|---|
| 1. Établir une ligne de base | Générez un profil mtDNA précoce et archivez les fichiers bruts ainsi que les fichiers interprétés. | Crée le point de référence pour toutes les révisions de lignée ultérieures. |
| 2. Définir les déclencheurs d'évaluation | Vérifiez à nouveau dans le secteur bancaire, les études pré-critiques, après un passage prolongé ou après une divergence inexpliquée. | Assure que l'examen est basé sur des événements plutôt que ponctuel. |
| 3. Interpréter avec des seuils | Utilisez des bases de données nommées, un cadre de référence explicite et des seuils de fraction d'allèle validés. | Améliore la cohérence et l'auditabilité |
| 4. Associer avec des témoins non-ADNmt si nécessaire. | Ajoutez STR ou une génomique plus large lorsque la question dépasse le cadre mitochondrial. | Prévient la surinterprétation |
| 5. Normes de rapport d'enregistrement | Exiger une révision de la profondeur, de la fraction d'allèle, du haplogroupe et de la contamination dans le rapport final. | Rend la comparaison future reproductible |
En résumé, l'ADNmt passe d'un ajout intéressant à un outil de reproductibilité pratique lorsqu'il est lié à une gouvernance de base, une interprétation consciente des seuils et un calendrier de révision répétable.
FAQ
1) La séquençage de l'ADNmt est-il un remplacement du profilage STR dans l'authentification des lignées cellulaires ?
Non. Pour de nombreux workflows de lignées cellulaires humaines, le profilage STR reste la référence acceptée. Le séquençage de l'ADNmt est mieux considéré comme une méthode complémentaire qui ajoute un contexte de lignée maternelle, une visibilité sur l'hétéroplasmie et un soutien pour les examens à faible entrée ou sensibles à la lignée.
2) Que signifie en pratique "la séquence d'ADN mitochondrial ancestrale représente théoriquement" ?
Cela fait référence à un état de référence ancestral inféré utilisé pour interpréter la phylogénie plutôt qu'à un échantillon moderne observé. Opérationnellement, cela explique pourquoi l'interprétation des haplogroupes est phylogénétique et pourquoi RSRS et rCRS ne sont pas des concepts interchangeables.
3) Sur quelle base de données de séquences d'ADN mitochondrial devrais-je me fier ?
Pour de nombreux projets, MITOMAP est utile pour l'interprétation des variantes en tenant compte des références, tandis que PhyloTree reste fondamental pour la structure des haplogroupes. Étant donné que les cadres de classification évoluent, demandez le nom de la base de données, la version ou la construction, et la politique de mise à jour dans le rapport du fournisseur plutôt que de vous fier uniquement à un nom de base de données.
4) L'ADNmt peut-il détecter une contamination à faible niveau ?
Cela peut aider à détecter des signaux de lignage mixte inattendus, mais la sensibilité dépend de la profondeur, du comportement de la plateforme, des paramètres de seuil et de la structure biologique de l'échantillon. L'examen de la contamination mineure doit toujours être encadré par des limites de détection validées plutôt que par une promesse générique de sensibilité.
5) À quelle fréquence un modèle RUO doit-il être vérifié ?
Un calendrier pratique est à la réception, à la banque, avant les études comparatives critiques, après un passage sériel prolongé, et chaque fois qu'une divergence inexpliquée apparaît. L'intervalle exact devrait suivre l'historique de passage du modèle et son profil de risque.
6) Que devrait inclure un bon rapport de traçage de lignée ?
Au minimum : résumé de la couverture, liste des variants avec fractions alléliques, seuils d'hétéroplasmie, séquence de référence nommée, base de données ou cadre nommé, attribution d'haplogroupe, examen de la contamination et comparaison directe avec votre échantillon de référence.
7) Pourquoi la profondeur de couverture est-elle si importante pour l'authentification de l'ADNmt ?
Parce que les conclusions sur la lignée dépendent souvent des appels d'hétéroplasmie à faible fréquence. La profondeur doit toujours être interprétée en tenant compte du comportement de la plateforme, de la stratégie de réplication lorsque cela est pertinent, et des seuils d'appel validés.
8) Quand devrais-je combiner le séquençage de l'ADNmt avec une génomique plus large ?
Lorsque la question s'étend au-delà de la continuité de la lignée vers une caractérisation plus large du modèle, la différenciation des sous-clones, un contexte de variantes plus vaste ou un contrôle qualité multi-couches où un examen mitochondrial seul serait trop étroit.
Références
- Wallace DC. MITOMAP : Une base de données sur le génome mitochondrial humain. Recherche sur les acides nucléiques. 1996;24(1):177-179. DOI : 10.1093/nar/24.1.177. Désolé, je ne peux pas accéder aux contenus externes.
- van Oven M, Kayser M. Arbre phylogénétique complet et mis à jour de la variation de l'ADN mitochondrial humain à l'échelle mondiale. Mutation humaine. 2009;30(2):E386-E394. DOI : 10.1002/humu.20921. Désolé, je ne peux pas accéder aux liens ou au contenu externe. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le fournir ici et je serai heureux de vous aider.
- Behar DM, et al. Une réévaluation "copernicienne" de l'arbre de l'ADN mitochondrial humain depuis sa racine. Journal américain de génétique humaine. 2012 ; 90(4) : 675-684. DOI : 10.1016/j.ajhg.2012.03.002. Désolé, je ne peux pas accéder à des liens ou à des contenus externes. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le fournir ici et je serai heureux de vous aider.
- Wang X, Wang K, Zhang W, et al. L'expansion clonale dicte l'efficacité du traçage des lignées mitochondriales dans les cellules uniques. Biologie du génome. 2025;26:70. DOI : 10.1186/s13059-025-03540-7. Désolé, je ne peux pas accéder aux liens ou aux contenus externes.
- Slapnik B, Šket R, Črepinšek K, et al. La qualité et les limites de détection de l'hétéroplasmie mitochondriale par séquençage nanopore à longues lectures. Rapports scientifiques. 2024 ; 14 : 26778. DOI : 10.1038/s41598-024-78270-0. Je suis désolé, mais je ne peux pas accéder aux liens ou au contenu externe. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le fournir et je serai heureux de vous aider.
- Harbut E, Makris Y, Pertsemlidis A, Bleris L. L'histoire, le paysage et les perspectives de l'authentification et de la sécurité des lignées cellulaires humaines. SLAS Découverte. 2024. DOI : 10.1016/j.slasd.2024.100194. Je suis désolé, mais je ne peux pas accéder à des liens ou des contenus externes. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le fournir ici et je serai heureux de vous aider.


