
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Dépannage du protocole de séquençage de l'ADNmt : surmonter les défis de la matrice d'échantillon dans la recherche B2B
Les projets de séquençage de l'ADN mitochondrial (ADNmt) semblent souvent simples car le génome est compact, circulaire et présent en plusieurs copies par cellule. Dans les flux de recherche externalisés réels, cette simplicité se dégrade rapidement. Le rapport ADNmt:ADN nucléaire (ADNn) varie selon les matrices, les intrants dégradés modifient ce qui peut réellement être enrichi, et les segments d'ADN mitochondrial nucléaire (NUMTs) peuvent fausser le mapping, la révision de l'hétéroplasmie et l'interprétation de la couverture, à moins que le flux de travail ne soit conçu pour les gérer dès le départ. La PCR à longue portée, la capture par sonde et l'amplification par cercle roulant (RCA) peuvent toutes bien fonctionner, mais elles ne s'adaptent pas aux mêmes conditions d'échantillon ou échouent de la même manière.
Pour les équipes B2B, la question opérationnelle n'est pas simplement de savoir si l'ADNmt peut être séquencé. La question plus utile est de savoir quel flux de travail peut générer des données de recherche adaptées aux besoins pour la prise de décision interne sur les projets, en respectant les contraintes de matrice convenues, les règles de contrôle qualité, le calendrier et le périmètre d'analyse. C'est pourquoi le choix du protocole, l'optimisation spécifique à la matrice, la bioinformatique consciente des NUMT et l'évaluation des fournisseurs devraient être considérés comme un processus de planification connecté plutôt que comme des transferts séparés.
La complexité de la sélection des protocoles de séquençage de l'ADN mitochondrial
Le séquençage de l'ADNmt n'est pas simplement un séquençage de petit génome. La première complication est le décalage d'abondance. Le nombre de copies d'ADNmt varie selon le type d'échantillon et le contexte d'extraction, tandis que l'ADN nucléaire peut dominer l'entrée totale même lorsque la question de recherche est purement mitochondriale. Cela signifie que la même masse nominale d'ADN peut se comporter très différemment selon les projets, surtout lorsque la fraction utilisable de molécules mitochondriales intactes est faible.
La deuxième complication est l'interférence des NUMT. Les NUMT sont des fragments dérivés de l'ADNmt intégrés dans le génome nucléaire. Parce qu'ils peuvent conserver une forte similarité de séquence avec l'ADNmt véritable, ils peuvent être co-amplifiés en laboratoire ou mal assignés lors du mapping. Les conséquences pratiques sont des variants faussement positifs, des signaux de faible fréquence gonflés, des estimations d'hétéroplasmie instables et des motifs de couverture qui semblent techniques mais sont en réalité interprétatifs. La conception du laboratoire humide à elle seule n'est pas suffisante ; des règles de filtrage et de reporting bioinformatique doivent être planifiées à l'avance.
PCR longue portée vs. capture par sonde vs. RCA
PCR à longue portée est souvent la voie la plus propre lorsque l'échantillon contient des molécules mitochondriales suffisamment intactes. Son principal avantage est la spécificité : un amplicon long correctement conçu peut réduire le transfert de NUMT et produire un enrichissement ciblé fort. Sa limitation est la fragilité. Une fois que l'intégrité de l'ADN diminue, le succès de l'amplification devient moins prévisible. La PCR à long terme unique a été mise en avant comme une méthode pratique pour éviter l'interférence omniprésente des NUMT lorsque des modèles intacts sont disponibles.
Capture de sonde est généralement plus tolérant aux entrées fragmentées car il ne nécessite pas la récupération de modèles presque complets avant l'enrichissement. Cela le rend attrayant pour les matrices dégradées, mais cela augmente également l'importance de la conception des sondes et du filtrage en aval, car des fragments nucléaires homologues peuvent encore être récupérés. La capture résout donc un problème tout en augmentant le besoin d'une stratégie de gestion des ambiguïtés clairement documentée.
RCA peut être utile lorsque l'enrichissement par modèle circulaire est avantageux et que l'entrée est limitée. Dans l'étude MitoRS, l'enrichissement basé sur l'RCA a soutenu l'amplification de l'ensemble du mitogénome en une seule réaction, a été décrit comme plus facile à mettre en place que l'amplification PCR classique, et a été associé à des paramètres ajustés pour l'analyse de l'hétéoplasmie à faible fréquence. Néanmoins, l'RCA n'est pas une méthode de secours universelle ; son succès dépend encore de la composition de l'entrée, du comportement de la bibliothèque et du filtrage en aval.
Une règle de décision pratique est simple. Utilisez la PCR à long terme lorsque l'intégrité est élevée et que l'objectif est un enrichissement propre du génome mitochondrial complet. Orientez-vous vers la capture lorsque la fragmentation est substantielle et que la récupération à partir de modèles endommagés est plus importante que la continuité intacte des longs modèles. Envisagez l'RCA lorsque l'entrée est limitée, que l'enrichissement de modèles circulaires est avantageux et que le fournisseur peut expliquer comment le contrôle de la révision à faible fréquence est effectué. Pour les programmes qui pourraient par la suite s'étendre au-delà de l'ADNmt, il peut être utile d'aligner la voie d'enrichissement avec des domaines adjacents. Séquençage de région ciblée flux de travail.
 Figure 1. Arbre de décision basé sur une matrice pour sélectionner la PCR à longue portée, la capture par sonde ou la RCA dans les projets mtDNA RUO.
Figure 1. Arbre de décision basé sur une matrice pour sélectionner la PCR à longue portée, la capture par sonde ou la RCA dans les projets mtDNA RUO.
Quand utiliser le séquençage de l'ensemble du mitogénome, et quand ne pas le faire.
Utilisez le séquençage complet du mitogénome lorsque le projet nécessite une large découverte de variantes, une interprétation tenant compte des haplotypes ou une visibilité à l'échelle du génome sur la molécule circulaire. Ne vous y fiez pas par défaut lorsque la question de recherche est étroite, que la matrice est fortement compromise ou que le budget et le délai sont mieux adaptés à un design ciblé. Dans ces cas, un design plus étroit. Service de séquençage de panneaux génétiques ou Services de séquençage d'amplicons La méthode peut être plus opérationnelle que d'imposer un flux de travail de génome complet que l'échantillon ne peut pas supporter.
Optimisation Spécifique aux Échantillons : Des FFPE aux Matrices à Cellules Uniques
Les matrices difficiles ne échouent pas pour une seule raison. Les entrées similaires à FFPE sont dominées par la fragmentation et la modification chimique. Les échantillons à très faible entrée sont dominés par le biais d'amplification, l'inflation des duplicatas et le dropout stochastique. Les matériaux micro-input dérivés des cellules peuvent sembler acceptables par concentration mais sous-performer encore parce que la fraction mitochondriale utilisable est incohérente. Le changement de planification le plus utile est de cesser de se demander si un protocole est "bon" et de commencer à se demander quel mode de défaillance il est réellement conçu pour contrôler.
Matériau dégradé : la logique des courts fragments surpasse la logique idéalisée des longueurs complètes.
L'ADN dérivé de FFPE nécessite un état d'esprit de qualité différent de celui de l'ADN frais et de haute intégrité. La fragmentation associée au formol et la modification chimique peuvent se transmettre à la préparation de la bibliothèque et à l'analyse si le flux de travail n'est pas adapté. Les revues larges sur le séquençage FFPE recommandent de traiter le fardeau de dommages comme une véritable variable analytique plutôt que de se fier uniquement à l'entrée totale. Pour les projets d'ADNmt, cela signifie généralement éviter une hypothèse rigide selon laquelle des molécules de pleine longueur sont récupérables, redéfinir la logique des amorces vers des régions récupérables plus courtes lorsque cela est nécessaire, et accepter la reconstruction basée sur le chevauchement lorsque l'utilisation d'un seul modèle intact est irréaliste.
Échantillons à faible entrée : le contrôle du biais est plus important que la sensibilité nominale.
Les flux de travail à faible entrée semblent souvent réussis avant que leurs limites ne deviennent évidentes. Une bibliothèque peut être produite, mais des pics de profondeur, des abandons locaux, des empilements lourds en doublons ou des appels à basse fréquence instables peuvent encore rendre l'interprétation faible. La RCA peut fonctionner avec des entrées limitées, mais le succès à faible entrée n'est significatif que lorsque le fournisseur définit des plages d'entrée minimales et recommandées, le comportement attendu de la bibliothèque et le seuil pour la ré-extraction, la ré-enrichissement ou le re-séquencement.
Une question utile lors de l'examen des fournisseurs n'est pas seulement "Quel est le minimum requis ?" mais aussi "À quoi ressemble une bibliothèque à faible apport acceptable, et que se passe-t-il si la complexité est médiocre ?" Cette distinction est bien plus importante que les revendications de sensibilité nominale. Si un flux de travail est présenté comme adapté à des matrices de recherche mtDNA difficiles, ces limites devraient être explicites plutôt qu'impliquées dans une définition. Flux de travail de séquençage de l'ADNmt.
Logique de protocole basée sur des matrices recommandée
Un tableau de décision compact est plus utile qu'un paragraphe de meilleures pratiques générique car il permet de comparer les fournisseurs plus rapidement et clarifie ce qui constitue un flux de travail correspondant dans des conditions d'utilisation à des fins de recherche.
| Matrice | Modèle d'intégrité | Itinéraire préféré | Risque principal | Contrôle qualité requis avant le lancement | Action de secours |
|---|---|---|---|---|---|
| ADN extrait de haute qualité | Principalement intact, faible fragmentation | PCR à longue portée | Échec de long amplicon si une fragmentation cachée est présente. | Montant d'entrée, résumé d'intégrité, méthode d'extraction précédente | Passer à un carrelage ou une capture plus courte. |
| ADN de tissu de type FFPE ou dégradé | Fragmenté, chimiquement modifié | Capture de sonde ou amplicons à court chevauchement | Récupération inégale, bruit associé aux dommages | Comportement des fragments, historique des dommages, attentes de récupération des cibles | Re-conception pour des fragments récupérables plus courts |
| Matériau cellulaire à très faible apport | Masse limitée, complexité moléculaire variable | RCA ou enrichissement ciblé court | Inflation dupliquée, abandon, appels à basse fréquence instables | Plage d'entrée recommandée et minimale, critères de complexité, déclencheur de rééchantillonnage | Répétition de l'extraction ou passage à un test plus étroit. |
| Échantillons dérivés de cellules nécessitant une confiance d'identité | Entrée variable, spécifique au lot | Dépendant de la matrice ; privilégier un enrichissement robuste ainsi que des contrôles d'identité. | Confusion entre échantillons croisés, mauvaise comparabilité | Note de chaîne de custody, plan de contrôle d'identité, cohérence de la matrice | Ajouter une vérification d'identité orthogonale |
| Question de recherche confirmatoire étroite | Adéquat ou limité | Itinéraire d'amplicon ciblé | Dépenses excessives pour des données de génome complet inutiles | Loci définis, périmètre de reporting, attente de délai de traitement. | Développez plus tard uniquement si les résultats initiaux le justifient. |
Les projets nécessitant une confiance en l'identité inter-échantillons dans les matériaux dérivés des cellules devraient associer la rigueur des protocoles à authentification de lignées cellulaires.
Liste de contrôle pour la soumission d'échantillons
Utilisez la liste de contrôle suivante avant l'expédition de l'échantillon afin que le fournisseur évalue la même définition de projet que vous :
| Élément de pré-soumission | Ce qu'il faut documenter |
|---|---|
| Type de matrice | Tissu, culot cellulaire, ADN extrait, archive dégradée ou autre matrice de recherche |
| Méthode d'extraction | Comment l'ADN a été isolé et si des étapes de nettoyage ou de réparation ont été utilisées. |
| Résumé de fragment ou d'intégrité | Comportement de fragmentation approximatif ou évaluation de l'intégrité |
| Objectif du projet | Découverte complète du mitogénome, examen confirmatoire ou examen ciblé à faible fréquence. |
| Revue à faible fréquence nécessaire | Oui ou non, avec des limites de reporting attendues. |
Cette même discipline de pré-lancement aide également lorsque le projet comprend des étapes orthogonales telles que Séquençage de Sanger pour confirmation de spot ou Test de quantification du nombre de copies d'ADN mitochondrial pour le contexte de l'abondance.
Conception de flux de travail en bioinformatique : cartographie, filtrage des NUMT et révision des variants
En pratique, le cartographie de l'ADNmt et l'analyse de séquence déterminent si les données d'enrichissement restent interprétables. Même lorsque l'enrichissement semble réussi, l'étape computationnelle doit encore séparer les lectures mitochondriales réelles des alignements ambigus ou dérivés de NUMT, gérer correctement le génome circulaire et appliquer des seuils qui correspondent à la profondeur du projet, au profil d'erreur et à l'objectif de révision.
La qualité de cartographie n'est pas un indicateur cosmétique.
La qualité de l'alignement est une variable de contrôle, pas une note de bas de page. Un alignement de faible confiance est souvent le premier signe que le flux de travail mélange la véritable séquence d'ADNmt avec une séquence nucléaire homologue ou des lectures mal localisées. Les revues axées sur les NUMT notent que le filtrage par qualité de séquence, fréquence des variants et contexte peut réduire les faux positifs, mais précisent également qu'aucune règle unique ne résout le problème. En pratique, un pipeline robuste combine stratégie d'alignement, filtrage des ambiguïtés, examen local des sites suspects et reporting contextuel.
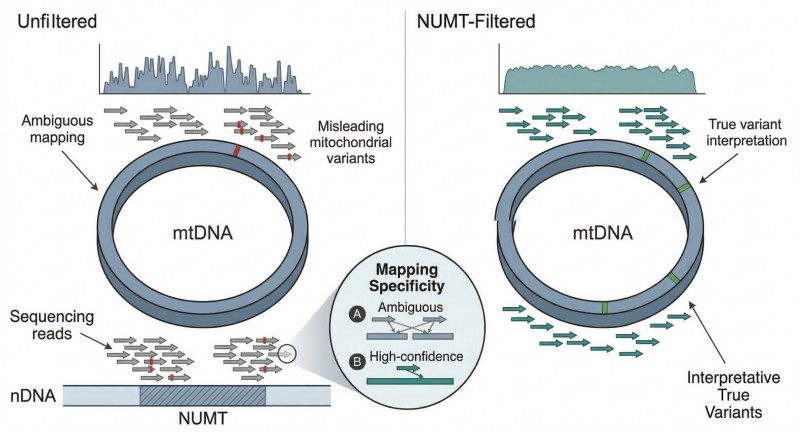 Figure 2. Comment le filtrage conscient des NUMT améliore la révision de la cartographie de l'ADNmt.
Figure 2. Comment le filtrage conscient des NUMT améliore la révision de la cartographie de l'ADNmt.
La figure distingue le placement de lecture ambigu des signaux à haute confiance conservés après révision de référence combinée et filtrage des ambiguïtés.
Optimisation du rapport signal/bruit dans la revue de l'hétéroplasmie
L'hétéroplasmie à faible fréquence est l'une des principales raisons pour lesquelles les équipes choisissent le séquençage profond de l'ADN mitochondrial, mais c'est aussi là que commence la surinterprétation. Les travaux publiés montrent que les flux de travail à haut débit peuvent détecter l'hétéroplasmie à faible fréquence, tout en soulignant que les signaux de faible niveau sont sensibles à la manipulation des données, à la contamination, à l'interférence des NUMT et au choix du seuil. C'est pourquoi un seuil universel unique est rarement approprié pour tous les matrices et flux de travail.
Une approche RUO pratique consiste à séparer seuils de dépistage de seuils de déclarationLe dépistage peut rester suffisamment permissif pour conserver des sites potentiels à faible fréquence pour examen. Le rapport devrait nécessiter des preuves plus solides, telles que le soutien des brins, la confiance dans le mapping, l'examen du contexte local et la cohérence avec les règles de QC et d'examen prédéfinies. Cette distinction est particulièrement importante dans les matrices endommagées ou à faible entrée, où la charge d'artefacts est rarement uniforme à travers le génome.
Problèmes courants en bioinformatique et comment les trier
Un tableau de triage rend cette section plus actionnable pour les équipes d'approvisionnement, de gestion de projet et d'examen technique en séparant le bruit récupérable des problèmes limitant le projet.
| Symptôme | Cause probable | Premier contrôle | Action d'escalade | Note de rapport final |
|---|---|---|---|---|
| Couverture inégale à travers le mitogénome | Biais d'amplicon, entrée dégradée, déséquilibre de capture | Couverture par région, limites des amplicons, comportement de duplication | Rebalancez la conception d'enrichissement ou réorganisez uniquement si la matrice le permet. | Notez si les lacunes sont générées par la matrice ou par la conception. |
| Variantes à basse fréquence suspectes dans des régions récurrentes | Report de NUMT ou cartographie ambiguë | Qualité de cartographie, alignement de référence combiné, support de brin | Appliquer un filtrage d'ambiguïté plus strict et une révision manuelle du site. | Marquer comme non résolu si une ambiguïté subsiste. |
| Artefacts près du carrefour circulaire | Gestion de référence linéarisée ou effets de bord | Rotation de référence, remappage conscient des jonctions | Reprocess avec une logique consciente du génome circulaire | Les appels associés aux jonctions ont été examinés séparément. |
| Haute profondeur mais faible confiance dans les appels | Inflation de profondeur basée sur des duplicatas ou faible complexité | Comportement unique des fragments, taux de duplication, complexité de la bibliothèque | Réextraire, réenrichir ou restreindre le périmètre de reporting | Précisez que la profondeur nominale n'était pas égale au soutien indépendant. |
La littérature publiée sur l'hétéroplasmie de l'ADNmt et les NUMT soutient ce type de revue en couches, car une profondeur apparente ne garantit pas l'interprétabilité.
Prise de décision stratégique pour les chefs de projet B2B
Pour les équipes B2B, le meilleur protocole d'ADNmt n'est pas celui qui semble le plus avancé dans une proposition. C'est celui qui correspond clairement au comportement de la matrice, à un contrôle qualité réalisable, à la transparence de l'analyse et à l'utilité des livrables. Les acheteurs et les examinateurs techniques n'ont pas besoin d'évaluer exactement les mêmes critères, mais ils devraient travailler à partir de la même définition de projet écrite.
Coût, délai de traitement et intégrité des données : un cadre pratique
Choisissez le séquençage de l'ensemble du mitogénome lorsque le projet nécessite une large découverte de variants, une visibilité des délétions ou un contexte mtDNA à l'échelle du génome. Optez pour une approche plus étroite lorsque la question est limitée, que la matrice est trop compromise pour un rétablissement fiable de la longueur complète, ou que le délai et le budget comptent plus que l'exhaustivité. L'erreur principale est de payer pour une exhaustivité que l'échantillon ne peut pas soutenir. Dans l'évaluation BOFU, un flux de travail plus étroit mais fiable est souvent préférable à un flux de travail large qui sera ensuite qualifié par de multiples réserves.
Table d'évaluation des fournisseurs
Transformez l'évaluation des fournisseurs en un tableau réutilisable au lieu d'une liste de questions génériques.
| Catégorie | Que demander | Pourquoi c'est important | Réponse à drapeau rouge |
|---|---|---|---|
| Limites d'entrée | Quelles sont les plages d'entrée minimales et recommandées par matrice ? | Empêche les incohérences de protocole avant l'approbation du bon de commande. | "Nous évaluerons cela plus tard après le début du séquençage." |
| Justification de l'itinéraire | Pourquoi la PCR, la capture ou l'RCA sont-elles recommandées pour cette matrice ? | Indique si le flux de travail est basé sur un échantillon ou piloté par un modèle. | "Ceci est notre flux de travail par défaut pour tous les projets d'ADNmt." |
| Contrôle NUMT | Comment les NUMTs sont-ils minimisés à la fois dans l'enrichissement et le mapping ? | Réduit les faux positifs et les appels d'hétéroplasmie instables. | "Les NUMTs ne posent généralement pas de problème majeur." |
| paquet QC | Quels sont les indicateurs de qualité (QC) inclus avant la bibliothèque, pendant la bibliothèque et après l'exécution ? | Précise les normes d'acceptation avant la livraison. | "Nous ne rapportons que le résultat final du séquençage." |
| Logique de seuil | Comment les sites à basse fréquence sont-ils filtrés et rapportés ? | Prévenir la confusion des seuils et l'interprétation excessive. | "Nous utilisons un seuil standard pour chaque échantillon." |
| Actions de secours | Qu'est-ce qui déclenche la ré-extraction, la ré-enrichissement ou le re-séquençage ? | Définit la responsabilité lorsque des matrices difficiles sous-performent. | "Nous gérons les échecs au cas par cas après l'arrivée des résultats." |
| Livrables | Quels fichiers bruts et traités, résumés et notes de méthode sont inclus ? | Rend les transferts et les revues en aval reproductibles. | "Nous fournissons uniquement un résumé." |
Normes d'acceptation à documenter avant le lancement
Au minimum, notez le parcours d'enrichissement convenu, les critères de rejet des échantillons, la logique de couverture utilisable attendue, le traitement des doublons ou de la complexité, la politique de révision des faibles fréquences et les formats de fichiers de livraison avant le début du séquençage. Cela réduit les litiges en aval car le projet est jugé sur des critères d'utilisation en recherche prédéfinis plutôt que sur des revendications de débit génériques. Utilisez cette liste de contrôle avant l'approbation de la commande pour que l'acceptation des échantillons, les actions de secours et la portée des rapports soient convenues par écrit.
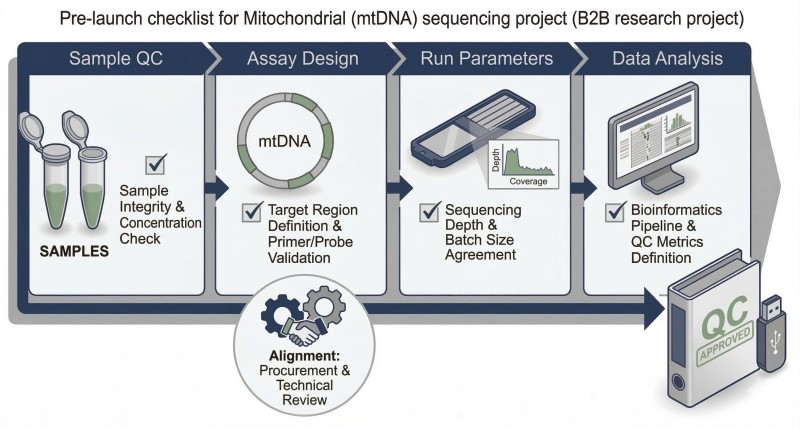 Figure 3. Liste de vérification pré-lancement pour l'échantillon, le contrôle qualité, l'analyse et l'alignement des livrables.
Figure 3. Liste de vérification pré-lancement pour l'échantillon, le contrôle qualité, l'analyse et l'alignement des livrables.
Utilisez cette liste de contrôle avant l'approbation du bon de commande afin que l'acceptation des échantillons, les actions de secours et le périmètre de reporting soient convenus par écrit.
Résumé : Anticiper l'avenir de votre recherche mitochondriale
Le séquençage de l'ADNmt continuera à s'améliorer à mesure que la conception d'enrichissement, l'examen ultra-profond et les flux de travail à longues lectures élargissent ce qui peut être résolu à travers le génome mitochondrial. La leçon la plus durable tirée de la littérature actuelle n'est pas qu'une seule plateforme résout tout, mais que l'enrichissement conscient de la matrice et l'analyse consciente des NUMT restent essentiels, quelle que soit le choix de la plateforme. La préparation à l'avenir commence par une gestion pré-analytique standardisée, un ajustement réaliste entre la matrice et la voie d'enrichissement, et un flux de travail de rapport qui indique clairement à la fois les résultats de contrôle qualité et les limitations d'utilisation à des fins de recherche.
Pour les lecteurs prévoyant un travail d'interprétation en aval, voir Analyse comparative de la fonction mitochondriale. Pour les programmes qui pourraient par la suite s'étendre au-delà de l'ADNmt, un adjacent Séquençage du génome entier Le flux de travail peut être une étape plus naturelle que de disperser son attention trop tôt sur des types d'essais non liés.
FAQ
Quelle est la plus grande erreur dans la sélection du protocole d'ADNmt ?
Choisir la voie par habitude plutôt que par matrice. L'ADN de haute intégrité, l'ADN similaire à celui des FFPE et l'ADN à ultra faible entrée ne créent pas le même profil de risque, donc ils ne devraient pas être contraints dans un même design d'enrichissement.
2) Pourquoi les NUMTs posent-ils tant de problèmes dans le séquençage de l'ADN mitochondrial ?
Parce que les NUMTs peuvent ressembler suffisamment aux lectures mitochondriales réelles pour fausser le mapping, la révision à faible fréquence et l'interprétation des variants si l'enrichissement et le filtrage ne sont pas conçus pour les contrôler.
3) La PCR à long rayon d'action est-elle toujours le meilleur choix pour le séquençage de l'ensemble du mitogénome ?
Non. C'est souvent excellent pour les modèles intacts, mais l'ADN dégradé peut rendre l'amplification de longs modèles peu fiable. Dans ces matrices, les stratégies de capture ou de chevauchement plus courtes sont généralement plus robustes.
4) Quand est-ce que l'RCA a du sens ?
RCA est le plus utile lorsque l'enrichissement par modèle circulaire est avantageux, que l'entrée est limitée et que le fournisseur peut expliquer comment le contrôle des revues à faible fréquence est géré en aval.
5) Comment un fournisseur doit-il rapporter les seuils d'hétéroplasmie ?
Pas comme un simple nombre isolé. Les seuils devraient être liés à la profondeur, à la confiance dans la cartographie, aux critères de révision et au risque d'artefact spécifique à la matrice.
6) Quels livrables un projet mtDNA B2B devrait-il inclure ?
Au minimum, les résultats de séquençage bruts, les fichiers d'alignement traités, les résumés de couverture, les tableaux de variants ou d'hétéoplasmie, les métriques de contrôle qualité et une note méthodologique concise expliquant la logique d'enrichissement et de filtrage.
7) Les matrices difficiles peuvent-elles encore soutenir des projets utiles sur l'ADNmt ?
Oui, mais seulement lorsque le flux de travail est repensé autour de la matrice plutôt que traité comme un échantillon standard. Les matériaux dégradés et à faible entrée nécessitent des hypothèses d'enrichissement et de contrôle qualité différentes.
8) Quand un acheteur devrait-il choisir un test plus étroit plutôt que le séquençage complet du mitogénome ?
Lorsque la question de recherche est ciblée, l'échantillon est trop compromis pour une récupération fiable du génome complet, ou l'élargissement des données n'améliorerait pas la décision réelle du projet.
Références
- Steiert TA, Weissensteiner H, Kronenberg F, et al. Un éclairage critique sur les paradigmes du séquençage de l'ADN FFPE. Recherche sur les acides nucléiques2023 ; 51(14) : 7143-7162. DOI : 10.1093/nar/gkad519. Désolé, je ne peux pas accéder à des liens externes ou à des contenus spécifiques. Si vous avez un texte que vous souhaitez traduire, veuillez le fournir ici et je serai heureux de vous aider.
- Smith AL, Hua H, Jensen-Seaman MI. Le puissant NUMT : l'ADN mitochondrial flexe son code dans le génome nucléaire. Biomolécules2023 ; 13(5) : 753. DOI : 10.3390/biom13050753. Désolé, je ne peux pas accéder aux liens ou au contenu externe. Si vous avez un texte spécifique à traduire, veuillez le fournir ici.
- Emser SV, Schaschl H, Millesi E, Steinborn R. Extension de l'enrichissement du mitogénome basé sur la PCR longue portée unique : ADNmt et peptides dérivés des mitochondries présumés de cinq hibernants rongeurs. Frontières en Génétique. 2021;12:685806. DOI : 10.3389/fgene.2021.685806. Je suis désolé, mais je ne peux pas accéder à des liens ou des contenus externes. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le copier ici et je serai heureux de vous aider.
- Gould MP, Bosworth CM, McMahon S, et al. MitoRS, une méthode pour la détection à haut débit, sensible et précise de l'hétéroplasmie de l'ADN mitochondrial. BMC Genomics. 2017;18:326. DOI : 10.1186/s12864-017-3695-5. Désolé, je ne peux pas accéder aux liens ou aux contenus externes. Si vous avez un texte spécifique à traduire, veuillez le fournir ici et je serai heureux de vous aider.
- Li M, Schönberg A, Schaefer M, Schroeder R, Nasidze I, Stoneking M. Détection de l'hétéroplasmie à partir du séquençage à haut débit des génomes mitochondriaux humains complets. Le Journal américain de génétique humaine2010 ; 87(2) : 237-249. DOI : 10.1016/j.ajhg.2010.07.014. Désolé, je ne peux pas accéder à des liens ou des contenus externes. Si vous avez un texte spécifique à traduire, veuillez le fournir ici et je serai heureux de vous aider.
- Liu Y, Schröder J, Schmidt B. Capture de séquences d'ADN multiplexées des génomes mitochondriaux à l'aide de produits PCR. PLOS ONE2010 ; 5(11) : e14004. DOI : 10.1371/journal.pone.0014004. Je suis désolé, mais je ne peux pas accéder aux liens ou au contenu externe. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le fournir ici et je serai heureux de vous aider.
- Arellano S, Wang L, Walzer K, et al. Séquençage hautement multiplexé et efficace de longs amplicons PacBio et Nanopore des génomes mitochondriaux de centaines à des milliers d'échantillons. BMC Genomics. 2023;24:190. DOI : 10.1186/s12864-023-09277-6. Désolé, je ne peux pas accéder à des liens ou à des contenus externes. Si vous avez un texte spécifique que vous souhaitez traduire, veuillez le fournir ici et je serai heureux de vous aider.


