
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Analyse comparative des séquences d'ADNmt : déchiffrer la divergence fonctionnelle mitochondriale dans les modèles de recherche
La comparaison des séquences mitochondriales devient difficile non pas lorsque les variants sont difficiles à détecter, mais lorsque les résultats deviennent trop faciles à surinterpréter. Dans de nombreux projets RUO, les équipes disposent déjà de lectures brutes acceptables, d'une liste de variants et d'un tableau d'hétéroplasmie, mais la question suivante reste sans réponse : Que montre réellement la comparaison des séquences d'ADN mitochondrial ? Cela nécessite une comparaison structurée à travers les régions génomiques, les classes de mutations, le niveau d'hétéroplasmie, le contexte de conservation et la qualité des rapports. Le résultat ne devrait pas se limiter à une liste de différences ; il devrait s'agir d'un cadre d'interprétation classé qui reste compatible avec l'analyse en aval et la révision des livrables par les fournisseurs. L'analyse de l'ADNmt présente également plusieurs contraintes techniques bien connues, notamment le choix de la séquence de référence, l'interférence des NUMT et la sensibilité au seuil pour l'hétéroplasmie à faible fréquence, de sorte que l'interprétation biologique doit toujours être liée au contrôle de qualité et aux hypothèses du pipeline.
Comparé aux workflows à l'échelle du génome plus larges, la comparaison de l'ADN mitochondrial (mtDNA) a une cible génomique plus étroite mais un fardeau d'interprétation plus nuancé. Le génome mitochondrial est compact, riche en gènes et fonctionnellement couplé à la phosphorylation oxydative, de sorte qu'un petit nombre de différences bien classées peut être plus informatif qu'une longue liste non triée. En même temps, chaque différence n'est pas nécessairement significative sur le plan fonctionnel. Les changements dans les régions codantes doivent être séparés des variations dans les régions de contrôle, les substitutions synonymes des candidats non synonymes, et les motifs stables spécifiques à un groupe des appels bruyants à faible niveau. Pour les projets de recherche B2B, cette distinction est importante car le véritable livrable n'est pas seulement des données de séquence, mais un récit analytique prêt à la décision : quelles variantes méritent un suivi, quelles différences peuvent refléter un contexte de lignée plutôt qu'une divergence fonctionnelle, et quels signaux restent provisoires en raison de préoccupations liées à la profondeur, à la référence ou à la contamination.
La question centrale : Que montre la comparaison des séquences d'ADN mitochondrial ?
Au niveau le plus basique, l'analyse comparative des séquences d'ADNmt devrait répondre à quatre questions : où se situent les différences, quel type de changements de séquence il s'agit, à quelle fréquence ils se produisent au sein et entre les groupes, et si le modèle observé est plausible en tant qu'hypothèse fonctionnelle plutôt qu'en tant que simple marqueur de lignée. Une bonne comparaison commence donc par partitionner le génome en loci codant pour des protéines, loci d'ARNr/ARNt, et le D-boucle/région de contrôleLes changements dans les régions codantes entraînent généralement le premier tour de priorisation fonctionnelle, en particulier pour les gènes contribuant aux complexes OXPHOS, tandis que les différences dans la boucle D sont plus souvent interprétées dans le contexte de la régulation, des signaux associés à la réplication ou de la discrimination entre groupes, plutôt que comme un effet immédiat au niveau des protéines. Cette distinction évite une erreur précoce et courante : traiter toutes les différences de l'ADNmt comme équivalentes.
La deuxième couche est la classe de mutation. Un cadre de révision utile sépare les changements synonymes, les substitutions non synonymes, les petites indels et les variants non codants. En pratique, la liste des candidats de première passe priorise généralement les substitutions non synonymes dans des positions conservées, suivies par des changements non codants récurrents qui sont fortement associés à des groupes et techniquement robustes. Cela ne signifie pas que les changements synonymes ou dans les régions de contrôle doivent être ignorés ; cela signifie qu'ils ne devraient pas occuper le même niveau d'interprétation que les candidats altérant les acides aminés jusqu'à ce que des preuves supplémentaires soient disponibles. Une manière pratique de présenter cela dans un rapport est d'annoter chaque site avec le contexte génomique, la conséquence sur les acides aminés, la profondeur, l'estimation de l'hétéroplasmie et un simple indicateur de priorisation. Ce format est beaucoup plus facile à examiner pour un propriétaire d'analyse en aval qu'un extrait brut de VCF seul.
La troisième couche est l'hétéroplasmie. Dans les projets comparatifs, le même locus peut se comporter très différemment selon que l'allèle alternatif est proche de l'homoplasmie, modérément hétéroplasique, ou observé seulement à faible fraction. L'hétéroplasmie n'est pas une métrique décorative ; elle modifie la confiance avec laquelle on doit interpréter les différences entre groupes. Un site présent à une profondeur élevée et avec une hétéroplasmie reproductible à travers les répliques est analytiquement différent d'un appel à faible fréquence limite près du seuil de détection. Des évaluations récentes montrent que la sensibilité et la fiabilité de la détection de l'hétéroplasmie dépendent fortement de la profondeur, de la technologie et du profil d'erreur, surtout lorsque l'analyse tente de résoudre des mélanges à faible niveau. Pour cette raison, l'interprétation intergroupe est la plus solide lorsque les distributions d'hétéroplasmie sont comparées ensemble avec la profondeur de lecture, l'équilibre des brins et la reproductibilité, et non comme des pourcentages isolés.
 Figure 1. Paysage comparatif des variantes d'ADNmt entre les groupes de recherche.
Figure 1. Paysage comparatif des variantes d'ADNmt entre les groupes de recherche.
Une carte mtDNA circulaire mettant en évidence les régions codantes, le D-loop, la densité des hotspots spécifiques au groupe et l'intensité de l'hétéroplasmie afin que les lecteurs puissent distinguer les sites candidats de haute priorité des variations de fond.
Une règle de révision pratique est la suivante : la comparaison devrait montrer motif, pas seulement présenceSi la sortie ne prouve que l'existence de variantes, l'analyse est incomplète. Si elle montre que certains loci sont enrichis dans un groupe de recherche, que les changements de codage candidats se regroupent dans des régions fonctionnellement pertinentes, et que les motifs d'hétéroplasmie sont systématiquement décalés entre les groupes, alors la comparaison commence à soutenir une hypothèse de divergence fonctionnelle adaptée à un suivi RUO. L'objectif n'est pas de revendiquer un mécanisme trop tôt, mais d'établir un chemin classé allant de la différence de séquence à la question expérimentale.
Flux de travail pour l'analyse avancée des séquences d'ADN mitochondrial
Un flux de travail avancé de comparaison de l'ADNmt commence généralement par un examen des entrées standardisées. Pour l'évaluation des livrables du fournisseur, cela signifie vérifier que le package du projet comprend des fichiers FASTQ propres, des fichiers BAM prêts pour l'alignement ou pré-alignés, un fichier de variantes ou un ensemble d'appels tabulaire, et un résumé de contrôle de qualité suffisamment détaillé. Pour un examen de style M-02, le Q30 à lui seul n'est pas suffisant ; le package doit également préciser la profondeur moyenne de l'ADNmt, la fraction ciblée si une enrichissement a été utilisé, le comportement de duplication et la logique de calcul de l'hétéroplasmie. La compatibilité du pipeline s'améliore considérablement lorsque les conventions de nommage, la construction de référence et le schéma d'annotation sont explicites dès le départ. C'est également à ce stade que la qualité des méthodes en amont est importante : une interprétation comparative de haute confiance dépend de la génération de données adaptée à l'objectif, que l'étude soit construite autour de flux de travail de séquençage de l'ADNmt ou plus ciblé séquençage d'ampliconsLes lecteurs travaillant à rebours à partir des problèmes d'analyse devraient également revoir Optimisation du protocole de séquençage de l'ADNmt pour des échantillons complexes avant d'attribuer des motifs ambigus en aval à la biologie plutôt qu'à la conception en amont.
Après la révision des entrées, l'alignement multi-séquence et la gestion des références deviennent le prochain point de contrôle. Pour les comparaisons axées sur l'humain, la séquence de référence de Cambridge révisée reste une base commune pour le reporting de position et l'harmonisation des annotations, et MITOMAP continue de fonctionner comme une ressource interprétative actuelle pour la recherche de locus, la recherche d'allèles, l'analyse de séquences basée sur MITOMASTER et le contexte de position lié à rCRS. Dans les modèles RUO pertinents pour l'humain, la cohérence avec les coordonnées basées sur rCRS soutient une comparaison inter-études plus claire. Dans les projets non humains ou inter-espèces, cependant, la portabilité des coordonnées à elle seule n'est pas suffisante ; l'analyse doit explicitement séparer la "commodité de référence humaine" de la "comparaison biologique appropriée à l'espèce". Cela signifie généralement aligner d'abord au sein des espèces, puis utiliser une interprétation consciente de la conservation pour l'inférence fonctionnelle inter-espèces.
Pour les pipelines automatisés ou semi-automatisés, des outils tels que MToolBox sont précieux car ils font plus que simplement assembler ou appeler des variants. Ils prennent également en charge une sortie consciente de l'hétéoplasmie et une logique de priorisation, ce qui est utile lorsque le réviseur a besoin de plus qu'une liste plate de sites. Un flux de travail solide comprend généralement un contrôle de qualité des lectures, un alignement ou une assemblage axé sur l'ADNmt, un filtrage conscient des NUMT, une évaluation de la couverture, un appel de variants conscient de l'hétéoplasmie, une annotation et une couche de priorisation finale. Les NUMT méritent une attention particulière ici. Les fragments mitochondriaux intégrés dans le noyau peuvent déformer le signal apparent de l'ADNmt si le flux de travail ne les contrôle pas, que ce soit dans la conception en laboratoire humide ou dans le filtre computationnel. En termes pratiques, tout signal diffus à faible fraction, surtout s'il se répète dans des échantillons non liés, devrait déclencher une vérification des NUMT avant que l'interprétation biologique ne s'élargisse. Lorsque la réutilisation en aval est importante, une structure pipeline d'appel de variants et compatible séquençage de bibliothèque préfabriquée les stratégies ne sont utiles que lorsque les hypothèses de référence, de filtrage et d'annotation sont suffisamment transparentes pour une réutilisation interne.
La prédiction fonctionnelle est mieux appliquée comme une couche de priorisation, et non comme un substitut à l'évidence. Pour les candidats non synonymes dans les gènes codant des protéines de l'ADNmt, des outils tels que SIFT et PolyPhen-2 peuvent aider à classer les substitutions en fonction de leur impact structurel ou fonctionnel probable. Cela est particulièrement utile lorsque la comparaison donne lieu à de nombreux changements d'acides aminés et que l'équipe a besoin d'une liste restreinte pour un examen plus approfondi. Néanmoins, ces outils doivent être utilisés pour un tri comparatif plutôt que pour une inflation des conclusions : un changement sur un site conservé plus un score de dommage prédit plus un enrichissement de groupe cohérent équivaut à un candidat de suivi plus solide, et non à une déclaration finale sur le mécanisme.
Interpréter la divergence fonctionnelle dans les contextes de recherche B2B
Dans les modèles de recherche orientés vers le métabolisme, une séquence interprétative utile est : détecter des variants spécifiques à un groupe, prioriser par contexte génomique et conservation, estimer les conséquences protéiques probables, puis relier les candidats les plus solides à des résultats de recherche mesurables tels que l'efficacité associée à l'ATP, l'équilibre redox ou les différences de réponse au stress. La formulation est importante. Dans les contextes RUO, la bonne conclusion est généralement "soutient une hypothèse de comportement fonctionnel mitochondrial altéré" plutôt que "prouve un effet défini." Les équipes qui souhaitent ensuite une confirmation orthogonale peuvent étendre la comparaison avec quantification du nombre de copies de l'ADNmt ou avec un plus large flux de travail d'intégration multi-omiques, en fonction de l'étendue du projet.
La comparaison inter-espèces ajoute un autre filtre interprétatif : la conservation. Une position qui est stable chez des organismes apparentés mais modifiée chez un modèle peut mériter plus d'attention qu'un changement à un site naturellement variable. C'est là que de nombreux articles deviennent trop centrés sur l'humain. Si le projet compare des modèles non humains, l'analyse ne devrait pas dépendre uniquement de la commodité des coordonnées humaines. Au lieu de cela, elle devrait intégrer un alignement sensible aux espèces et un scoring de conservation, et elle devrait clairement indiquer quand une conclusion est soutenue par la conservation inter-espèces plutôt que par des ressources d'annotation uniquement humaines. Cette distinction est souvent la différence entre un rapport comparatif convaincant et un rapport difficile à défendre lors d'une révision technique.
Une matrice de priorisation pratique pour la divergence fonctionnelle inclut généralement la région génomique, la classe de mutation, la plage d'hétéroplasmie, le contexte de conservation, l'impact protéique prédit si applicable, l'enrichissement entre groupes et l'étape suivante recommandée.
Pour rendre l'interprétation comparative opérationnelle, le rapport doit passer d'une prose descriptive à une matrice de priorisation fixe. Dans les environnements RUO, chaque site candidat doit être examiné selon les mêmes critères : région génomique, classe de mutation, comportement d'hétéroplasmie, contexte de conservation, confiance technique, enrichissement entre groupes et action recommandée. Cela évite le problème courant de surpondération des variants visuellement frappants qui manquent de reproductibilité ou de contexte. Cela facilite également la comparaison des livrables externalisés avec les pipelines internes, car la logique de classement devient explicite plutôt que dépendante de l'évaluateur. Pour la plupart des projets, une sortie en trois niveaux est suffisante : candidats à suivi de haute priorité, différences contextuelles mais de moindre priorité, et appels provisoires nécessitant une révision QC avant que l'interprétation ne s'élargisse.
Matrice de priorisation prête à décider
| Site/Région | Classe de Mutation | Modèle d'hétéroplasmie | Conservation | Confiance technique | Enrichissement de groupe | Niveau de priorité | Étape suivante recommandée RUO |
|---|---|---|---|---|---|---|---|
| Locus codant conservé | Non-synonyme | Déplacement reproductible entre les réplicats | Élevé | Profondeur élevée, soutien équilibré | Fort | Élevé | Revue de l'impact des protéines plus suivi orthogonal ciblé |
| Locus de codage | Synonyme | Stable mais modeste | Modéré | Élevé | Modéré | Moyen | Conservez comme signal contextuel ; ne menez pas l'interprétation seul. |
| point chaud du D-loop | Non-codant | Signal spécifique à un groupe fort | Variable | Élevé | Fort | Moyen | Traiter comme un candidat associé à la réglementation ou à la lignée ; valider la cohérence. |
| Toute région | N'importe quelle classe | Proche du seuil ou instable | Tout | Profondeur faible ou soutien incohérent | Flou | Provisoire | Vérifiez à nouveau le contrôle qualité, le filtrage et le contexte de l'échantillon avant l'interprétation. |
| Appels récurrents de faible niveau à travers des échantillons non liés | N'importe quelle classe | Diffuse | Tout | Suspect | Faible | Provisoire | Examine d'abord la gestion des NUMT et la spécificité de l'alignement. |
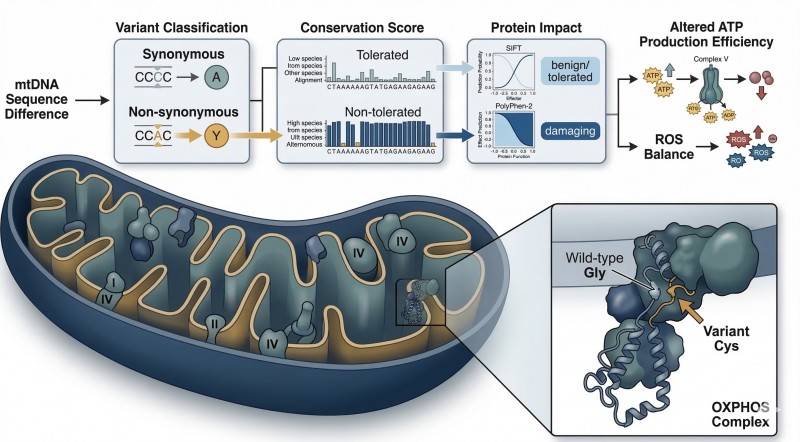 Figure 2. Des différences de séquence de l'ADNmt aux hypothèses de divergence fonctionnelle.
Figure 2. Des différences de séquence de l'ADNmt aux hypothèses de divergence fonctionnelle.
Une carte logique en couches reliant les différences de séquence à la prédiction de l'impact sur les protéines et aux hypothèses au niveau des voies impliquant le comportement de l'OXPHOS, la production associée à l'ATP et les observations de recherche liées aux ROS.
Une manière utile de présenter le reste de l'interprétation est sous la forme d'un cadre "quoi observer ensuite".
Catégories de caractéristiques mitochondriales courantes et points d'observation de recherche
Gènes OXPHOS codant pour des protéines
Priorisez les substitutions non synonymes, en particulier aux résidus conservés. Observez les essais liés à l'ATP, les résultats associés au redox ou les lectures liées au potentiel de membrane dans le système modèle.
loci d'ARNr et d'ARNt
Interprétez avec prudence mais ne négligez pas. Ceux-ci peuvent influencer l'efficacité mitochondriale liée à la traduction et méritent une annotation structurée plutôt qu'un rejet par défaut. MITOMAP et MITOMASTER restent utiles pour le contexte de position de séquence, tandis que les ressources d'annotation élargies ont amélioré le soutien à l'interprétation des mt-tRNA.
D-loop / région de contrôle
Le mieux évalué pour le contrôle de réplication, le contexte réglementaire et la séparation des groupes, en particulier lorsqu'il est répété à travers des échantillons avec un fort soutien technique. Une sur-interprétation est courante ici, à moins que la différence ne soit stable et clairement enrichie.
Surmonter les goulets d'étranglement de l'analyse : des listes de variants aux insights biologiques
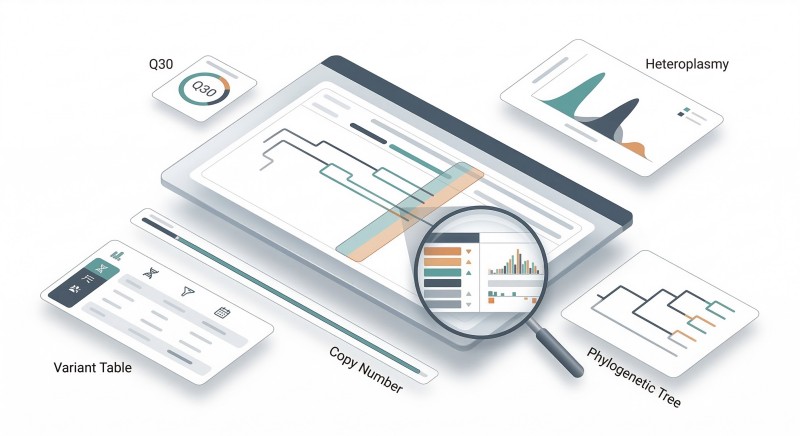 Figure 3. Rapport d'analyse comparative de l'ADNmt : Des métriques de contrôle qualité à la priorisation biologique.
Figure 3. Rapport d'analyse comparative de l'ADNmt : Des métriques de contrôle qualité à la priorisation biologique.
Un tableau de bord de style rapport montrant la combinaison des métriques de CQ, de la distribution de l'hétéroplasmie, du regroupement phylogénétique, de l'annotation des variants et du contexte du nombre de copies nécessaire pour une analyse comparative prête à la décision.
Le goulot d'étranglement le plus courant dans les travaux comparatifs sur l'ADNmt n'est pas la détection des variants. C'est l'écart entre un ensemble d'appels techniquement acceptable et une liste courte biologiquement utile. Lorsque le rapport contient des dizaines ou des centaines de différences, les équipes ont souvent besoin d'une seconde couche d'interprétation qui combine la variation de séquence avec le contexte de l'échantillon et la confiance analytique. C'est pourquoi les meilleurs rapports comparatifs sont des sorties multi-panneaux plutôt que des tableaux uniques. Ils devraient montrer un résumé compact de contrôle de qualité, une vue de la distribution d'hétéroplasmie, un tableau de variants candidats regroupés, et un résumé tenant compte des lignées qui aide à distinguer la structure ancestrale de fond des signaux de priorité fonctionnelle. Avant d'étendre l'interprétation, les équipes devraient confirmer l'identité de l'échantillon par identification de lignées cellulaires et examiner le contexte de lignée connexe à travers tracer la lignée mitochondriale dans l'authentification des lignées cellulaires RUO.
Un autre goulot d'étranglement est la relation faible entre le nombre de variants et l'insight biologique. Plus de variants ne signifient pas nécessairement plus de valeur explicative. Dans de nombreux projets, l'interprétation devient plus claire lorsque les différences de séquence de l'ADNmt sont examinées en conjonction avec le contexte du nombre de copies. Le nombre de copies de l'ADNmt ne remplace pas la comparaison de séquences, mais il peut ajouter une seconde dimension utile lorsque l'équipe souhaite savoir si un modèle diffère uniquement par la composition de la séquence ou également par l'abondance génomique mitochondriale. Même dans la littérature de recherche non clinique, les analyses du nombre de copies de l'ADNmt ont montré que l'abondance génomique peut changer de manière significative la façon dont les hypothèses sur l'état mitochondrial sont formulées.
QC et dépannage : symptôme → cause probable → prochaine action
Des nombres inattendus de variantes à faible fréquence dans de nombreux échantillons.
Cause probable : interférence NUMT, ambiguïté de cartographie ou seuils d'appel trop permissifs.
Prochaine action : inspecter la spécificité de l'alignement, re-vérifier la stratégie d'enrichissement de l'ADNmt, renforcer les filtres de l'appelant et examiner les régions sensibles aux NUMT connues.
Les estimations d'hétéroplasmie varient fortement entre les répétitions techniques.
Cause probable : profondeur insuffisante, comportement d'erreur spécifique à la plateforme ou prétraitement incohérent.
Prochaine action : vérifier la distribution de profondeur, le support de brin, la gestion des duplicatas et la définition exacte de l'hétéroplasmie utilisée dans le rapport. L'étude de référence sur les longues lectures de 2024 a rapporté un seuil de détection d'hétéroplasmie de 12 % à 150× de couverture dans son cadre évalué, ce qui rappelle utilement que les seuils de détection dépendent de la technologie et du flux de travail plutôt que d'être universels.
De nombreuses différences mais peu de structure de groupe interprétable.
Cause probable : mélange de lignées en arrière-plan, incohérence d'identité des échantillons ou variants de faible priorité dominant le rapport.
Prochaine action : appliquer un regroupement tenant compte de la lignée, prioriser les candidats de codage conservés et confirmer la cohérence des échantillons avant d'étendre l'interprétation fonctionnelle.
Signal D-loop fort mais signal de région codante faible
Cause probable : enrichissement de la région régulatrice, séparation des lignées, ou variation riche en région de contrôle sans conséquence directe sur les protéines.
Prochaine action : éviter de forcer un récit sur l'impact des protéines ; plutôt, positionner le résultat comme une découverte réglementaire ou de contexte comparatif, à moins que d'autres preuves ne le renforcent.
Bonne Q30 mais faible interprétabilité en aval.
Cause probable : livrables incomplets, annotation superficielle ou rapport non structuré.
Prochaine action : demander des données brutes et des sorties d'annotation standardisées, des informations de référence explicites et un tableau de variantes prêt pour la priorisation. Une bonne qualité de base ne compense pas un emballage interprétatif médiocre.
Conclusion : Renforcer les études RUO avec des analyses comparatives précises
L'analyse comparative des séquences d'ADNmt est particulièrement utile lorsqu'elle transforme la différence de séquence en un chemin d'interprétation classé et techniquement défendable. Pour les modèles de recherche RUO, cela signifie aller au-delà de la simple présence de variantes brutes et se demander où se situe la différence, quelle classe de changement elle représente, à quel point le signal d'hétéroplasmie est robuste, si le site est conservé et à quel point le résultat peut être facilement intégré dans un raisonnement biologique en aval. Une comparaison bien menée ne réduit pas tout à une seule étiquette "fonctionnelle". Au contraire, elle sépare les variations de fond probables, le signal associé à la lignée et les candidats dignes d'un suivi. Cette approche est particulièrement précieuse pour les équipes de recherche B2B qui ont besoin à la fois de rigueur analytique et de compatibilité avec les pipelines dans les livrables des fournisseurs.
En regardant vers l'avenir, les stratégies de lecture longue pourraient améliorer davantage l'interprétation complète des molécules, le contexte de phasage et le comportement de détection dans des scénarios d'hétéroplasmie difficiles, en particulier lorsque le projet nécessite plus de confiance autour de signaux complexes ou de faible niveau. Des évaluations récentes suggèrent que les approches de lecture longue sont prometteuses, mais leur comportement analytique doit encore être compris dans le contexte des limites de détection et des caractéristiques d'erreur, plutôt que d'être supposé supérieur dans chaque cas d'utilisation. En d'autres termes, de meilleures analyses comparatives viendront non seulement d'un plus grand séquençage, mais aussi d'un meilleur classement des preuves, de livrables plus clairs et d'un couplage plus étroit entre le contrôle qualité et l'interprétation.
FAQ
1) Que montre la comparaison des séquences d'ADN mitochondrial dans un projet RUO typique ?
Il montre généralement si les différences sont concentrées dans les régions codantes, les régions de contrôle ou les compartiments génomiques mixtes ; si les changements sont synonymes ou non synonymes ; et si les distributions d'hétéroplasmie diffèrent entre les groupes de recherche de manière techniquement cohérente. Le résultat le plus fort est un ensemble de candidats priorisés, et non simplement une liste de variantes plus longue.
2) La séquence de l'ADN mitochondrial humain est-elle toujours la bonne référence pour la comparaison ?
Non. Les projets axés sur l'humain bénéficient souvent d'un reporting basé sur rCRS pour la cohérence, mais les modèles inter-espèces ou non humains nécessitent une comparaison consciente des espèces et une interprétation basée sur la conservation plutôt que sur la commodité de référence humaine seule.
3) Comment un responsable en bioinformatique devrait-il examiner un rapport de comparaison de l'ADNmt sous-traité ?
Commencez par l'exhaustivité des livrables : sortie d'alignement FASTQ/BAM ou équivalente, tableau des variants annotés, estimations d'hétéroplasmie, détails de référence et métriques de contrôle qualité claires, y compris la profondeur. Ensuite, examinez la logique de classement des candidats et si le rapport sépare la variation de fond des sites prioritaires fonctionnels.
4) Q30 est-il suffisant pour évaluer la qualité des données pour la comparaison de l'ADNmt ?
Non. La Q30 est utile mais incomplète. La profondeur moyenne de l'ADNmt, la distribution des lectures, le soutien à l'hétéroplasmie et les hypothèses de filtrage sont tous importants pour l'interprétabilité, surtout lorsque des signaux à faible fréquence sont comparés entre les groupes.
5) Les différences d'hétéroplasmie peuvent-elles être interprétées directement comme une divergence fonctionnelle ?
Pas par eux-mêmes. L'hétéroplasmie soutient l'interprétation lorsqu'elle est techniquement robuste et alignée avec le contexte génomique, la conservation et la pertinence des sites candidats. C'est une couche dans la chaîne d'inférence, pas la chaîne entière.
6) Pourquoi les NUMTs sont-ils importants dans l'analyse des séquences mitochondriales ?
Parce que les fragments mitochondriaux intégrés dans le noyau peuvent imiter le signal de l'ADNmt et créer des appels de faible fraction trompeurs ou des artefacts récurrents si le filtrage est faible. Le contrôle des NUMT est important tant dans la conception expérimentale que dans l'analyse des données.
7) Quand le nombre de copies d'ADNmt doit-il être ajouté à une étude de comparaison ?
Lorsque le projet nécessite une seconde dimension au-delà de la composition des séquences, en particulier si l'équipe souhaite comparer l'abondance génomique mitochondriale avec la charge de variants ou le modèle d'hétéroplasmie. C'est le plus utile en tant que contexte complémentaire, et non en remplacement de l'analyse des séquences.
8) Qu'est-ce qui rend un rapport d'ADNmt compatible avec un pipeline ?
Système de coordonnées cohérent, séquence de référence explicite, formats de fichiers standard, champs d'annotation reproductibles et une structure de priorisation pouvant être réintroduite dans les flux de travail en aval sans reconstruction manuelle.
Références
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Réanalyse et révision de la séquence de référence de Cambridge pour l'ADN mitochondrial humain. Nature Génétique1999 ; 23(2) : 147. DOI : 10.1038/13779.
- Wallace DC, Lott MT, Brown MD, et al. MITOMAP : Une base de données sur le génome mitochondrial humain. Recherche sur les acides nucléiques1996 ; 24(1) : 177-179. DOI : 10.1093/nar/24.1.177.
- MITOMAP. MITOMAP : Une base de données sur le génome mitochondrial humain. Format de citation du site MITOMAP, 2023 ; les outils actuels incluent la recherche d'allèles et MITOMASTER, le site affichant les mises à jour récentes de la base de données. ressource MITOMAP.
- Calabrese C, Simone D, Diroma MA, et al. MToolBox : un pipeline hautement automatisé pour l'annotation de l'hétéoplasmie et l'analyse de priorisation des variants mitochondriaux humains dans le séquençage à haut débit. Bioinformatique2014 ; 30(21) : 3115-3117. DOI : 10.1093/bioinformatics/btu483.
- Weissensteiner H, Pacher D, Kloss-Brandstätter A, et al. Une collection complète d'annotations pour interpréter la variation de séquence dans les ARN de transfert mitochondriaux humains. BMC Bioinformatique2016 ; 17 : 420. DOI : 10.1186/s12859-016-1193-4.
- Adzhubei IA, Schmidt S, Peshkin L, et al. Un procédé et un serveur pour prédire les mutations de missense dommageables. Méthodes de la nature2010 ; 7(4) : 248-249. DOI : 10.1038/nmeth0410-248.
- Ng PC, Henikoff S. SIFT : Prédire les changements d'acides aminés qui affectent la fonction des protéines. Recherche sur les acides nucléiques2003;31(13):3812-3814. DOI : 10.1093/nar/gkg509.
- Sturk-Andreaggi K, Renshaw M, et al. Hétéroplasmie de l'ADNmt : Origine, Détection, Signification et Conséquences Évolutionnaires. Vie2021;11(7):633. DOI : 10.3390/life11070633.
- Dayama G, Emery SB, Kidd JM, Mills RE. Le paysage génomique des insertions mitochondriales nucléaires polymorphes chez l'homme. Recherche sur les acides nucléiques. 2014;42(20):12640-12649. DOI : 10.1093/nar/gku1038.
- Slapnik B, Šket R, Črepinšek K, Tesovnik T, Jenko Bizjan B, Kovač J. La qualité et les limites de détection de l'hétéroplasmie mitochondriale par séquençage nanopore à longues lectures. Rapports Scientifiques. 2024;14:26778. DOI : 10.1038/s41598-024-78270-0.
- Castellani CA, Longchamps RJ, Sun J, Guallar E, Arking DE. Penser en dehors du noyau : le nombre de copies de l'ADN mitochondrial dans la santé et la maladie. Mitochondrie. 2020;53:214-223. DOI : 10.1016/j.mito.2020.06.004. (Europe PMC)


