
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Meilleures pratiques pour concevoir une expérience de séquençage Cut&Tag
CUT&Tag (combinant les technologies de marquage et de séquençage) est une méthode d'analyse génomique très efficace, largement utilisée pour étudier les interactions protéine-ADN, l'épigénétique et de nombreuses autres questions biologiques. Pour garantir le succès des expériences CUT&Tag, il est essentiel de suivre les meilleures pratiques lors de la conception des expériences. Cet article présente ces meilleures pratiques pour aider les chercheurs à obtenir des résultats expérimentaux fiables et de haute qualité.
I. Principes fondamentaux et planification pré-expérimentale de la conception expérimentale
Correspondance entre le type de cible et le design expérimental
- Modifications des histones (par exemple, H3K27me3, H3K4me3) : Des anticorps de qualité ChIP à haute spécificité doivent être sélectionnés, et leur applicabilité dans l'espèce cible doit être validée. Par exemple, l'anticorps H3K27me3 de CST (Cell Signaling Technology) (numéro de catalogue #9733) a démontré son efficacité dans plusieurs études.
- Facteurs de transcription (par exemple, CTCF, STAT3) : Il est préférable d'utiliser des anticorps avec des anticorps secondaires pré-adsorbés pour réduire la réactivité croisée, et la spécificité des anticorps doit être validée par des expériences de knockout/knockdown.
- Études sur les cellules uniques : La viabilité cellulaire (viabilité >85%) et l'intégrité nucléaire (membrane nucléaire intacte sans rupture au microscope) doivent être évaluées, et le nombre initial de cellules doit être contrôlé entre 1 000 et 10 000 cellules.
Procédure de préparation d'échantillons standardisée
- Collecte et lavage des cellules :
- Cellules en suspension : Collectez directement par centrifugation (300×g, 5 min) et lavez deux fois avec du PBS.
- Cellules adhérentes : Après digestion à la trypsine, resuspendre dans du PBS pré-refroidi pour éviter les dommages mécaniques pouvant entraîner une fuite d'ADN.
- Fixation et perméabilisation :
- Expériences de modification des histones : Aucun réticulation croisée n'est nécessaire. Perméabilisez directement avec de la digitonine (1,5–3 μg/mL) pendant 10 minutes pour maintenir l'état natif de la chromatine.
- Expériences sur les facteurs de transcription : Aucun réticulage n'est requis. Comme pour les études de modification des histones, la perméabilisation doit être effectuée directement à l'aide de Digitonine. Cela préserve l'état natif de la chromatine, garantit une efficacité optimale des réactions in situ tant pour les anticorps que pour l'enzyme Tn5, et fournit ainsi des données avec un rapport signal sur bruit élevé.
- Extraction nucléaire :
- Lysez les membranes cellulaires à l'aide d'un tampon hypotonique (10 mM Tris-HCl, 10 mM NaCl) pour préserver les noyaux intacts.
- Confirmer la viabilité nucléaire >90% par coloration au bleu de trypan et observer l'uniformité de la morphologie nucléaire au microscope.
Pour un guide d'introduction plus détaillé sur le séquençage CUT&Tag, veuillez vous référer à "Qu'est-ce que le séquençage Cut&Tag ? Un guide complet pour débutants".
 Optimisation expérimentale de CUT&Tag (Abbasova L et al., 2025)
Optimisation expérimentale de CUT&Tag (Abbasova L et al., 2025)
Directives de gestion des types d'échantillons spéciaux
Échantillons de tissus (cellules non cultivées)
- Échantillonnage et préservation : Les tissus frais doivent être immédiatement congelés par flash dans de l'azote liquide (conserver à -80°C pendant ≤3 mois), ou intégrés dans de l'OCT puis congelés à -80°C (éviter les cycles de congélation-dégel répétés).
- Procédure de dissociation : Digérer avec de la collagénase IV (1 mg/mL) + DNase I (10 U/mL) à 37°C pendant 30 minutes, puis filtrer à travers un tamis de 70 μm pour obtenir une suspension de cellules uniques.
Échantillons FFPE (Tissu paraffiné clinique)
- Déswaxage et récupération : Déswaxage deux fois avec du xylène (10 minutes à chaque fois) → Hydratation par éthanol en gradient → Récupération des antigènes à 95°C pendant 20 minutes avec un tampon de citrate de sodium (pH 6,0).
- Notes clés : L'ADN des échantillons FFPE est sujet à la fragmentation ; réduire le temps de réaction de transposition à 3-5 minutes et éviter les fragments trop petits (<100 pb).
Échantillons rares (par exemple, cellules tumorales circulantes, CTC)
- Volume initial : Minimum 500 cellules. Les cellules vivantes doivent être enrichies à l'aide de billes magnétiques ConA (avec la concanavaline A) pour éviter la contamination de l'ADN provenant des cellules mortes.
- Mesures de prévention des pertes : Utilisez des tubes de centrifugation à faible adsorption tout au long du processus. Après incubation avec des billes magnétiques, resuspendre délicatement les cellules dans du PBS (≤5 fois).
II. Opération Raffinée des Étapes Expérimentales Clés
Incubation des anticorps et capture du signal
- Incubation avec l'anticorps primaire :
- Ratio de dilution de l'anticorps des histones : 1:100–1:200 (par exemple, CST #9733).
- Ratio de dilution de l'anticorps du facteur de transcription : 1:50–1:100 (par exemple, Abcam #ab150471).
- Conditions d'incubation : Incuber pendant la nuit (12 à 16 heures) à 4°C avec rotation ou à température ambiante (25°C) pendant 2 heures pour assurer une liaison adéquate des anticorps au site cible.
- Pontage d'anticorps secondaires :
- Utilisez un anticorps secondaire fusionné à la protéine A/G (par exemple, Jackson ImmunoResearch #115-005-003) et incubez à température ambiante pendant 1 heure pour améliorer le ciblage du complexe Tn5.
- Utilisez un tampon de lavage spécialisé pré-refroidi contenant de la digitonine pour maintenir l'état perméabilisé des noyaux et éliminer efficacement les anticorps liés de manière non spécifique.
Contrôle précis de la tagmentation
- Préparation de complexe enzymatique :
- Il est recommandé d'utiliser une enzyme de fusion pA-Tn5 pré-calibrée (comme Biotech Cat#TD902), avec une unité d'activité ≥50 U/μL.
- Système de réaction : 1× tampon de tagmentation (contenant 10 mM de MgCl₂), en évitant l'introduction de composants inhibiteurs tels que le SDS.
- Optimisation des conditions d'activation :
- Température : Incuber à 37°C pendant 5 à 10 minutes. Une incubation excessive entraînera une coupure excessive de l'ADN (fragments <50 pb).
- Résiliation : Ajouter de l'EDTA (20 mM) ou de la protéinase K (digérer à 55°C pendant 30 minutes) pour inactiver l'activité de l'enzyme Tn5.
Normalisation de la construction de bibliothèques et contrôle de la qualité
- Extraction d'ADN :
- Utilisez une purification par billes magnétiques (comme les billes AMPure XP) pour éliminer les transposases et les protéines non liées.
- Sélection de la taille des fragments d'ADN : Des fragments de 100 à 600 pb ont été récupérés par gel à l'aide du système BluePippin, garantissant un pourcentage de bibliothèque efficace >80 %.
- Optimisation de l'amplification par PCR :
- Sélection d'enzymes à haute fidélité : KAPA HiFi HotStart ReadyMix (taux d'erreur <0,02 %).
- Contrôle du nombre de cycles : 10 à 12 cycles (augmenter à 15 cycles pour de faibles quantités de départ) afin d'éviter une sur-amplification et un biais.
- Normes de contrôle de la qualité :
- Analyse des fragments : L'Agilent 2100 Bioanalyzer a montré un pic principal à 200-300 pb sans traînée significative.
- Qualité de séquençage : plateforme Illumina NovaSeq, score de qualité Phred moyen Q30 >90%, contenu en GC 40–60%.
III. Intégration multi-omiques et conception expérimentale à haut débit
Stratégies d'optimisation pour le traitement parallèle de plusieurs échantillons
- Application de plateforme d'automatisation :
- Utilisez des plaques à 96 puits combinées à un système de tri par billes magnétiques (tel que le Beckman Coulter Biomek i7) pour automatiser l'ensemble du processus d'incubation des anticorps, de lavage et de réactions de transposition, réduisant ainsi l'erreur humaine.
- Volume de réaction recommandé : 50 μL/échantillon, garantissant l'homogénéité des réactifs à de faibles volumes de départ.
- Principes de conception des codes-barres :
- Stratégie d'index à double extrémité : combinaison d'index i5/i7 (comme Illumina Nextera XT), distance de Hamming ≥3, évitant le saut d'index.
- Ajout d'échantillon de contrôle interne : Mettre en place un contrôle IgG et un contrôle d'entrée pour chaque lot afin d'évaluer le bruit de fond et de calibrer les pics.
Analyse conjointe des données multi-omiques
- Intégration ATAC-seq :
- Utilisez le chevauchement entre les données H3K4me3 de CUT&Tag et les régions ouvertes dans l'ATAC-seq pour identifier les promoteurs/amplificateurs actifs.
- Outils recommandés : HOMER (annotatePeaks.pl) pour l'annotation fonctionnelle, et la base de données Cistrome DB pour l'alignement avec les éléments régulateurs connus.
- Analyse d'association RNA-seq :
- L'analyse de dépistage génique différentiel (DESeq2, padj<0,05) et l'analyse d'intersection des pics CUT&Tag ont été utilisées pour construire un réseau d'interaction entre éléments régulateurs de gènes.
- Outils de visualisation : UCSC Genome Browser ou IGV pour afficher les résultats de co-localisation.
IV. Problèmes courants et solutions approfondies
| Problème | Analyse des causes | Solution | Cas de référence |
|---|---|---|---|
| FRiP < 5% | Faible efficacité de liaison des anticorps ou activité de transposase insuffisante | Vérifiez la spécificité des anticorps (en utilisant des lignées cellulaires knockout), optimisez le temps de réaction de transposition. | Cas BioTechPack : FRiP augmenté à 18 % après optimisation de l'anticorps H3K27me3 |
| Enrichissement TSS insuffisant | Lyse nucléaire incomplète ou perméabilisation excessive | Ajustez le temps de perméabilisation (5 à 10 minutes), utilisez une faible concentration de NP-40. | Solution iGeneBio : Perméabilisation par digitonine + Capture par billes magnétiques ConA |
| Biais d'amplification de bibliothèque | Nombre de cycles PCR inapproprié ou mauvaise qualité de l'ADN modèle | Optimiser le nombre de cycles (10 à 12 cycles), purifier l'ADN à une concentration >5 ng/μL. | Kit Novozyme TD901 : Q30 > 95 % après amplification sur 12 cycles |
| Effets de lot significatifs | Fluctuations dans les conditions expérimentales ou désordre dans la séquence de traitement des échantillons | Utilisez une plateforme automatisée, incluez des échantillons de contrôle dans le même lot. | Processus BioTechPack : plaque à 96 puits + tri par billes magnétiques, CV de lot < 5 % |
V. Flux de travail d'analyse de données détaillées
Traitement des données brutes
- Contrôle de qualité et filtrage : FastQC détecte les bases de faible qualité (qualité Phred <20) et la contamination par des adaptateurs, et effectue le rognage en utilisant Trimmomatic (paramètres : ILLUMINACLIP:TruSeq3-SE:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36).
- Affinage des données : Le fichier BAM a été trié et indexé à l'aide de samtools pour générer le fichier final pour l'analyse en aval. Remarque : Dans l'analyse des données CUT&Tag, l'étape de marquage des duplicats PCR basé sur les coordonnées (par exemple, en utilisant Picard MarkDuplicates) doit être omise.
Identification et annotation des pics
- Appel de pics MACS2 :
- Pour les pics 'étroits' (par exemple, les facteurs de transcription, H3K4me3) : utilisez macs2 callpeak -t treatment.bam -c IgG_control.bam -f BAMPE -g hs -q 0,05. Si vous travaillez avec des données à extrémité unique ou si une correction explicite pour le décalage Tn5 est nécessaire, les paramètres --nomodel --shift -75 --extsize 150 peuvent être utilisés.
- Pour les pics 'larges' (par exemple, H3K27me3, H3K9me3) : Le paramètre --broad doit être ajouté, avec un seuil plus détendu, par exemple : macs2 callpeak -t treatment.bam -c IgG_control.bam -f BAM -g hs --broad --broad-cutoff 0.1.
- Annotation fonctionnelle : Le package `ChIPseeker` annote les régions génomiques (promoteurs, exons, amplificateurs) et les corrèle avec des données d'expression génique (par exemple, la base de données GEO).
- Analyse et visualisation avancées :
- Enrichissement de motifs : HOMER (findMotifs.pl) ou MEME Suite identifie les séquences centrales des sites de liaison (par exemple, le motif conservé de CTCF : CCTCCTC).
- Analyse différentielle : DESeq2 filtre les pics différemment exprimés (padj < 0,05), et `clusterProfiler` effectue une analyse d'enrichissement GO/KEGG.
- Outils de visualisation : IGV génère des pistes de couverture et des cartes de chaleur pour montrer la distribution génomique des sites de liaison des protéines cibles.
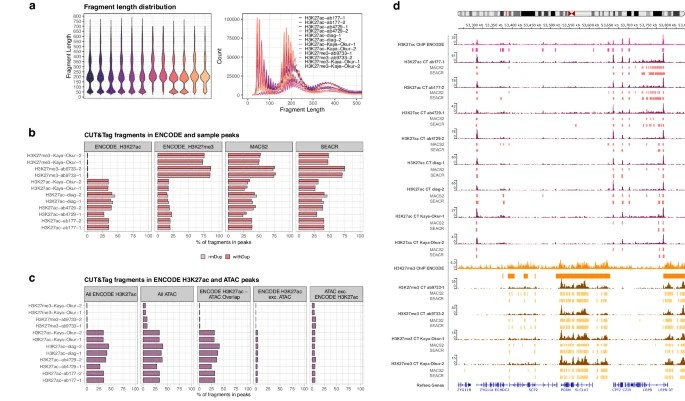 Métriques de contrôle de qualité pour les données CUT&Tag (Abbasova L et al., 2025)
Métriques de contrôle de qualité pour les données CUT&Tag (Abbasova L et al., 2025)
VI. Comparaison des avantages techniques et des limitations
| Avantages | Limitations | Contre-mesures |
|---|---|---|
| Exigence d'échantillon extrêmement faible (niveau de cellule unique) | Fortement dépendant de la qualité et de la spécificité des anticorps. Pour des cibles avec une abondance extrêmement faible, il peut être nécessaire d'optimiser les conditions expérimentales (par exemple, augmenter le nombre de cellules de départ, prolonger le temps d'incubation des anticorps) pour améliorer les taux de détection. | Valider le titre d'anticorps lors des pré-expériences ; enrichir les cibles fortement exprimées. |
| Bruit de fond faible (FRiP > 10%) | Impossible de distinguer les sites de liaison physiquement adjacents. | Intégrer des données ATAC-seq ou Hi-C pour résoudre les interactions tridimensionnelles. |
| Cycle expérimental court (réalisé en 24 heures) | Coût élevé pour les expériences sur cellules uniques | Adoptez des plateformes comme 10x Genomics pour réduire le coût par échantillon. |
VII. Protocole expérimental recommandé (en prenant le facteur de transcription CTCF comme exemple)
- Préparation des échantillons :
- Type de cellule : lignées cellulaires de cancer colorectal HCT116 (viabilité >90%).
- Volume initial : 5 000 cellules, lavées avec du PBS et resuspendues dans du PBS 1× + 0,1 % BSA.
- Incubation des anticorps :
- Anticorps primaire : anticorps CTCF (Abcam #ab150471), dilution 1:100, incubé toute la nuit à 4°C.
- Anticorps secondaire : IgG anti-lapin de chèvre (Jackson ImmunoResearch #111-005-003), dilution 1:200, incubé à température ambiante pendant 1 heure.
- Réaction de transposition : enzyme pA-Tn5, activée à 37°C pendant 8 minutes, ADN purifié après l'arrêt de la réaction.
- Construction de bibliothèque : Kit de préparation de bibliothèque d'ADN NEBNext Ultra II, amplification PCR 12 cycles, taille de fragment sélectionnée 200–400 pb.
- Séquençage et analyse :
- Plateforme : Illumina NovaSeq 6000, PE150, 15M lectures/échantillon.
- Flux de travail d'analyse : identification des pics MACS2 → analyse des motifs HOMER → annotation différentielle des pics.
VIII. Directions futures de développement technologique
- Intégration multi-omique à cellule unique : Combinaison de scCUT&Tag avec scATAC-seq pour élucider les dynamiques spatiotemporelles de l'accessibilité de la chromatine et de la liaison des protéines.
- Adaptation du séquençage par nanopore : Développement de schémas de construction de bibliothèques CUT&Tag compatibles avec le séquençage long-read pour capturer des éléments régulateurs complexes.
- Automatisation et assistance par IA : Optimisation des taux de succès expérimentaux en prédisant les conditions d'incubation des anticorps sur la base de l'apprentissage automatique.
Les gens demandent aussi
Comment fonctionne la tagmentation Tn5 ?
Illumina a développé le protocole de tagmentation, dans lequel une enzyme Tn5 modifiée coupe l'ADN double brin et ligature simultanément les séquences de liaison nécessaires pour le séquençage Illumina aux deux extrémités.
Quel est le mécanisme d'insertion de Tn5 ?
Tn5 se compose de deux séquences d'insertion IS50 qui encadrent trois gènes codant pour la résistance à la kanamycine, à la bléomycine et à la streptomycine. La transposition de Tn5 se produit par un mécanisme de coupe et collage, déplaçant le transposon du donneur vers la cible, sans créer de copies supplémentaires du transposon.
Quel est le processus de tagmentation ?
La tagmentation est la première étape de la préparation de la bibliothèque où l'ADN non fragmenté est clivé et étiqueté pour analyse. La préparation de bibliothèque par tagmentation sur billes utilise des transposomes liés à des billes pour une réaction de tagmentation plus uniforme par rapport aux réactions de tagmentation en solution.
Comment fonctionne la transposase Tn5 dans l'Atac Seq ?
Tn5 fragmente simultanément l'ADN, s'insère de manière préférentielle dans les sites de chromatine ouverte et ajoute des amorces de séquençage (un processus connu sous le nom de tagmentation). L'ADN séquencé identifie la chromatine ouverte et l'analyse des données peut fournir des informations sur la régulation des gènes.
Quel est le biais de la séquence de la transposase Tn5 ?
Le Tn5 présente un biais de séquence complexe qui n'est pas efficacement corrigé par les méthodes traditionnelles de correction de biais.
Références :
- Abbasova L, Urbanaviciute P, Hu D, Ismail JN, Schilder BM, Nott A, Skene NG, Marzi SJ. CUT&Tag récupère jusqu'à la moitié des pics d'acétylation des histones ChIP-seq d'ENCODE. Nat Commun. 2025 Mar 27;16(1):2993.
- Henikoff S, Henikoff JG, Ahmad K, Paranal RM, Janssens DH, Russell ZR, Szulzewsky F, Kugel S, Holland EC. Analyse épigénomique d'échantillons fixés au formol et inclus dans la paraffine par CUT&Tag. Nat Commun. 2023 22 sept.;14(1):5930.


