
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Marqueurs moléculaires d'ADN : dynamiques évolutives, découverte à haut débit et application stratégique dans la recherche génomique
Intention méta : Un guide stratégique et axé sur la recherche pour sélectionner des systèmes de marqueurs moléculaires ADN pour l'analyse de la diversité, le cartographie, les GWAS, la phylogénomique et les workflows de sélection.
Les marqueurs moléculaires d'ADN ont transformé la génomique en rendant la variation de séquence mesurable. Les premiers systèmes détectaient la variation de manière indirecte, à travers la taille des fragments ou les motifs de bandes. Les systèmes modernes résolvent de plus en plus les allèles à des loci définis et convertissent la variation en génotypes numériques portables. Ce changement a modifié plus que le débit. Il a changé la manière dont les chercheurs conçoivent des projets, comparent des ensembles de données et passent de la découverte générale à la validation en aval.
Cet aperçu est destiné à un usage de recherche et à la planification de flux de travail génomique uniquement, en mettant l'accent sur la sélection de marqueurs, la conception d'essais et la stratégie de données en aval.
La question la plus utile n'est plus de savoir quels systèmes de marqueurs existent. La vraie question est de savoir quel système correspond à l'objectif biologique. Un panel qui fonctionne bien pour l'analyse de la diversité peut être trop sparse pour le mapping fin. Un flux de travail de séquençage à représentation réduite qui est excellent pour la découverte de SNP peut être inefficace pour le génotypage routinier en aval. Un marqueur avec un polymorphisme générique élevé peut encore échouer en sélection si celui-ci ne suit pas de manière fiable le locus cible dans le germoplasme réel sous sélection. Pour cette raison, les marqueurs ADN sont mieux compris non pas comme une liste historique, mais comme une stratégie adaptée à un objectif.
 Figure 1. L'évolution des marqueurs reflète non seulement un débit plus élevé, mais aussi un passage des lectures de fragments à des appels de génotypes portables et définis par séquence.
Figure 1. L'évolution des marqueurs reflète non seulement un débit plus élevé, mais aussi un passage des lectures de fragments à des appels de génotypes portables et définis par séquence.
Le spectre des systèmes de marqueurs
Une façon pratique de classer les systèmes de marqueurs est par ce qu'ils détectent. Les anciennes plateformes détectent souvent des différences de longueur de fragments après digestion ou amplification. Les nouvelles plateformes détectent de plus en plus des états de séquence spécifiques à des positions génomiques définies. Cette distinction influence presque tout ce qui suit : l'interprétation des allèles, la reproductibilité, la portabilité entre laboratoires et la compatibilité avec les flux de travail statistiques modernes pour l'analyse de la diversité, le mapping ou les études d'association.
Des RFLP et AFLP aux SSR et SNP
La polymorphisme de longueur des fragments de restriction, ou RFLP, était l'un des premiers systèmes de marqueurs ADN robustes. Il repose sur la digestion par des enzymes de restriction suivie de la séparation des fragments et de la détection spécifique des loci. Les changements de séquence qui créent ou abolissent des sites de restriction, ou modifient la longueur des fragments, produisent des motifs différents. Le RFLP a été historiquement important car il était conscient des loci et reproductible, mais il était également lent, laborieux et difficile à mettre à l'échelle. Cela a limité sa valeur à long terme dans les études sur de grandes cohortes.
L'AFLP a conservé la logique de la digestion de restriction tout en augmentant la sortie multiplex. Il est devenu utile pour le fingerprinting et les travaux sur la diversité car de nombreux fragments polymorphes pouvaient être générés en un seul essai. Le compromis était l'interprétabilité. La présence et l'absence de fragments peuvent être informatives, mais elles ne préservent pas toujours la résolution complète des génotypes. À mesure que les systèmes basés sur le séquençage ont mûri, l'AFLP est devenu moins attrayant dans les projets nécessitant des génotypes portables et définis par locus.
Les SSR, ou microsatellites, ont marqué une avancée majeure car ils sont codominants et souvent très polymorphes. Au lieu d'évaluer un large motif de fragments, les chercheurs pouvaient comparer les allèles à des loci de répétition définis et distinguer directement les états homozygotes des états hétérozygotes. Pendant des années, les SSR ont été le cheval de bataille de la génétique des populations, de l'analyse de parenté, du cartographie de liaison et de l'évaluation des ressources génétiques. Leur valeur reste claire aujourd'hui. Ils fournissent souvent un contenu d'information élevé par locus, et leur nature multi-allélique peut être particulièrement utile dans des projets axés sur la diversité.
Les SNPs ont déplacé le domaine vers un mode de fonctionnement différent. La plupart des loci SNP ne sont que bi-alléliques, donc chaque locus individuel est généralement moins polymorphe qu'un SSR typique. Mais les SNPs sont abondants, largement distribués et hautement compatibles avec le génotypage basé sur des puces et le séquençage. Une fois qu'un grand nombre de SNPs pouvaient être évalués de manière économique et reproductible, l'analyse des marqueurs a évolué d'une maximisation du contenu d'information d'un locus vers une intégration des informations à travers des milliers de loci à l'échelle du génome. C'est une des raisons pour lesquelles les stratégies centrées sur les SNP dominent désormais la recherche génomique à haut débit.
Pourquoi les marqueurs codominants sont plus importants que les marqueurs dominants.
La différence entre les marqueurs dominants et codominants n'est pas un détail technique mineur. Elle détermine la quantité d'information biologique qui survit à l'analyse.
Un marqueur dominant réduit généralement les états génotypiques à la présence ou à l'absence. Dans les diploïdes, cela signifie qu'un hétérozygote peut devenir indistinguable d'une classe d'homozygote. Une fois cela arrivé, les estimations d'hétérozygotie, l'inférence des fréquences alléliques et l'analyse de la structure des populations deviennent toutes moins directes et souvent moins fiables.
Un marqueur codominant préserve les deux états alléliques à un locus. Les SSR le font généralement par des différences de longueur de fragments. Les SNP le font par des allèles définis par la séquence. Le résultat est une résolution génotypique plus claire et une meilleure compatibilité avec les analyses statistiques modernes.
C'est pourquoi les marqueurs codominants sont généralement préférés dans l'analyse de la diversité, l'estimation de la parenté, l'analyse d'admixture et la plupart des workflows de cartographie. Ils ne produisent pas seulement des données plus nettes. Ils préservent les classes de génotypes nécessaires pour mesurer l'hétérozygotie et modéliser correctement la ségrégation. En pratique, la codominance réduit la perte d'information au point exact où l'interprétation biologique commence.
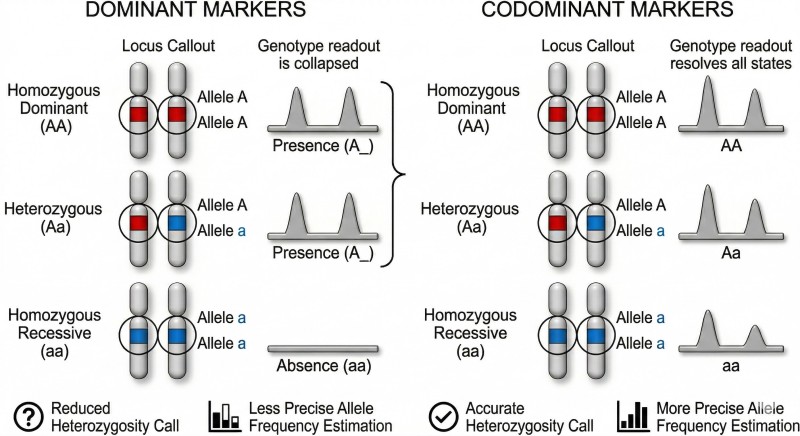 Figure 2. Les tests dominants compressent les états hétérozygotes et homozygotes, tandis que les marqueurs codominants préservent la résolution des génotypes pour l'analyse des fréquences alléliques, l'estimation de l'hétérozygotie et la structure des populations.
Figure 2. Les tests dominants compressent les états hétérozygotes et homozygotes, tandis que les marqueurs codominants préservent la résolution des génotypes pour l'analyse des fréquences alléliques, l'estimation de l'hétérozygotie et la structure des populations.
Le PIC et l'hétérozygotie sont liés, mais non interchangeables.
Deux termes apparaissent constamment dans l'évaluation des marqueurs : l'hétérozygotie et le contenu en information de polymorphisme, ou PIC. Ils sont liés, mais ce ne sont pas la même métrique.
L'hétérozygotie attendue décrit la probabilité que les allèles échantillonnés diffèrent à un locus. C'est une mesure de diversité influencée par les fréquences alléliques au sein de la cohorte cible. Le PIC dépend également des fréquences alléliques, mais il se concentre plus directement sur la manière dont le marqueur est informatif pour distinguer le polymorphisme dans cette population. Pour des distributions alléliques équilibrées, les deux valeurs tendent à augmenter. Pour des distributions alléliques fortement biaisées, les deux tendent à diminuer.
Cela a deux implications importantes. Premièrement, la qualité des marqueurs est spécifique à chaque cohorte. Le même marqueur peut être très informatif dans une population et faible dans une autre. Deuxièmement, un polymorphisme générique fort ne rend pas automatiquement un marqueur utile pour chaque projet. Un marqueur peut montrer un PIC élevé dans un panel de diversité tout en ayant de mauvaises performances en élevage s'il est faiblement lié à la région cible ou se comporte de manière incohérente entre les lignées d'élevage.
Pour le travail de découverte, une large informativité est importante. Pour la validation en aval, la qualité de liaison et la robustesse des tests sont souvent plus importantes. C'est pourquoi le PIC doit être considéré comme un indicateur utile, et non comme un score universel qui répond à chaque question de sélection de marqueurs par lui-même.
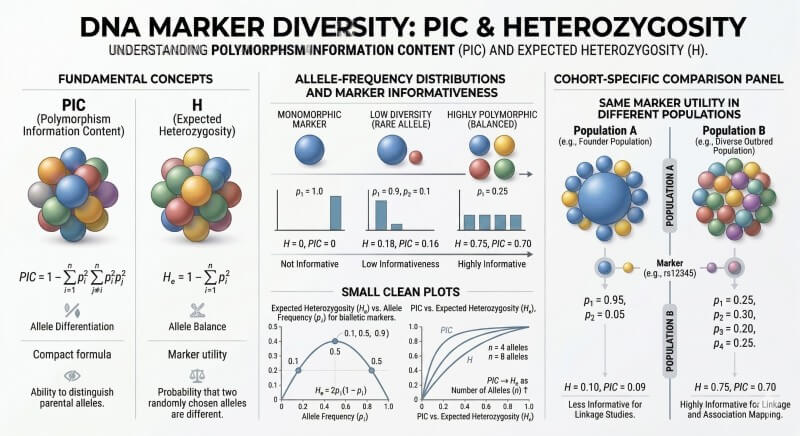 Figure 3. Le PIC et l'hétérozygotie dépendent tous deux de la distribution des fréquences alléliques, mais le PIC est spécifiquement utilisé pour évaluer l'informativeness des marqueurs au sein de la cohorte cible.
Figure 3. Le PIC et l'hétérozygotie dépendent tous deux de la distribution des fréquences alléliques, mais le PIC est spécifiquement utilisé pour évaluer l'informativeness des marqueurs au sein de la cohorte cible.
Souveraineté technique à l'ère des NGS
Le séquençage de nouvelle génération a changé la découverte de marqueurs en permettant aux chercheurs de découvrir et d'évaluer la variation dans le même cadre expérimental. Une fois que la sortie de séquençage est devenue évolutive, la question limitante n'était plus de savoir si le polymorphisme pouvait être détecté. La véritable question est devenue quelle fraction du génome devait être échantillonnée, comment elle pouvait être échantillonnée de manière reproductible entre les individus, et si l'ensemble de marqueurs résultant correspondrait à l'objectif en aval.
Les méthodes de séquençage à représentation réduite ont émergé de ce problème. Au lieu de séquencer chaque base, elles récupèrent intentionnellement des sous-ensembles génomiques reproductibles qui sont suffisamment riches pour la découverte de SNP et le génotypage. GBS, RAD-seq et DArTseq appartiennent tous à cette logique générale, mais ils résolvent le problème de différentes manières.
GBS : découverte de SNP à haut débit par réduction contrôlée de la complexité
Le génotypage par séquençage est souvent décrit comme une méthode SNP à faible coût, mais cette étiquette est trop superficielle. Le GBS fonctionne parce qu'il utilise des enzymes de restriction pour transformer un génome complet en une bibliothèque réduite reproductible. L'ADN génomique est digéré, des adaptateurs et des codes-barres sont ligaturés, les fragments sont regroupés, et la bibliothèque est séquencée en multiplex. Les fragments homologues récupérés chez différents individus peuvent ensuite être alignés et comparés pour la découverte de SNP et l'appel de génotypes.
La force du GBS réside dans la réduction contrôlée de la complexité. La méthode ne cherche pas à traiter chaque partie du génome de manière égale. Elle échantillonne le génome de manière stratégique. C'est pourquoi le choix des enzymes de restriction n'est pas un petit paramètre de protocole. C'est l'un des principaux déterminants de la qualité des données.
La fréquence des sites de reconnaissance affecte le nombre de fragments qui entrent dans la bibliothèque. La sensibilité à la méthylation influence si les régions génomiques répétitives, souvent moins informatives, sont sur-représentées ou supprimées. La taille du génome et la charge en répétitions déterminent combien de la bibliothèque résultante sera analytiquement utile. Un enzyme de restriction qui fonctionne bien dans un génome compact peut créer une complexité écrasante dans un grand génome riche en répétitions. Les symptômes en aval sont familiers : profondeur par locus peu profonde, récupération inégale des loci, données manquantes gonflées et filtrage plus lourd.
Le multiplexage ajoute une autre couche de compromis. Regrouper de nombreux échantillons est une des raisons pour lesquelles le GBS est rentable, mais le coût par échantillon ne diminue que tant que la profondeur de séquençage reste adéquate pour le design de l'étude. Une faible profondeur peut être acceptable dans certaines enquêtes à l'échelle de la population, surtout lorsque la densité des marqueurs est élevée et que l'imputation est réalisable. Cela devient beaucoup plus risqué lorsque la certitude du génotype individuel est importante, lorsque le génome de référence est faible, ou lorsque la structure de la population rend l'imputation instable. Des données bon marché ne sont pas toujours des données économiques.
Le cas d'utilisation le plus fort pour le GBS est la découverte génomique à grande échelle à un stade précoce. Il est très efficace pour la découverte de SNP larges, le cartographie de liaison dense, le dépistage de germoplasme et le travail exploratoire génotype-phénotype dans des cohortes trop grandes pour le séquençage de génome entier de routine. Il est souvent particulièrement adapté lorsque les projets nécessitent de nombreux marqueurs à travers de nombreux échantillons et peuvent tolérer un filtrage analytique.
Dans ce contexte, Génotypage par séquençage (GBS) est naturellement aligné avec des projets axés sur la découverte, tandis que Génotypage SNP du génome entier est souvent plus adapté lorsque des matrices SNP plus larges sont nécessaires pour de grands ensembles d'échantillons.
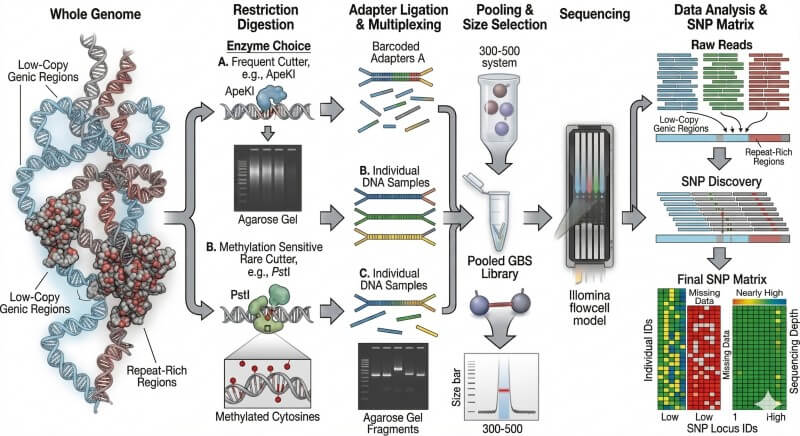 Figure 4. Le GBS réduit la complexité du génome par digestion de restriction et construction de bibliothèques multiplexées, mais le rendement des marqueurs et l'absence dépendent fortement du choix des enzymes, de la profondeur de séquençage et de l'architecture du génome.
Figure 4. Le GBS réduit la complexité du génome par digestion de restriction et construction de bibliothèques multiplexées, mais le rendement des marqueurs et l'absence dépendent fortement du choix des enzymes, de la profondeur de séquençage et de l'architecture du génome.
Pourquoi les projets GBS échouent lorsque la logique de conception est faible.
De nombreux résumés expliquent le flux de travail GBS mais s'arrêtent avant le véritable problème. Pourquoi certains ensembles de données GBS sous-performent-ils ?
La première raison est la récupération incohérente des fragments. Si la qualité de l'ADN varie, l'efficacité de la digestion change ou si la préparation de la bibliothèque introduit un biais, les mêmes loci peuvent ne pas être récupérés de manière uniforme entre les échantillons. La deuxième raison est une profondeur insuffisante. Si trop de fragments se disputent trop peu de lectures, la confiance dans le génotype diminue. La troisième raison est un soutien de référence insuffisant. Si le génome de référence disponible est fragmenté, éloigné ou incomplet, la qualité de l'alignement en souffre et l'interprétation des loci devient moins stable. La quatrième raison est un filtrage excessif après un design expérimental faible. Un projet peut commencer avec de nombreux loci candidats mais en perdre une grande partie une fois que les seuils de manque, de profondeur et de reproductibilité sont appliqués.
Ce ne sont pas des arguments contre le GBS. Ce sont des rappels que le GBS est une plateforme sensible au design. Les meilleurs projets de GBS ne sont pas construits à partir d'habitudes de protocole. Ils sont construits à rebours à partir du point de recherche.
RAD-seq : contexte de locus plus fort, mais compromis plus dépendants du protocole
Le RAD-seq appartient à la même famille de représentation réduite que le GBS, mais il ne doit pas être considéré comme interchangeable. Sa logique fondamentale est de récupérer et de séquencer l'ADN adjacent aux sites de restriction, ce qui fournit aux chercheurs un ensemble reproductible de loci flanquants pour la découverte de SNP et le génotypage. Cette structure a rendu le RAD-seq particulièrement influent dans la génomique écologique, les radiations phylogénétiques récentes, les études de divergence des populations et le fine mapping chez les organismes non-modèles.
La valeur du RAD-seq ne réside pas seulement dans le nombre de marqueurs. C'est la combinaison d'un large échantillonnage génomique avec un contexte de séquence local. Dans le bon cadre, cela le rend attrayant pour la phylogénomique à l'échelle des clades et le travail de cartographie ciblée. Mais le RAD-seq est très sensible à l'architecture du protocole. La perte de sites de restriction peut réduire la récupération des loci à travers des taxons divergents. Les choix de sélection de taille peuvent modifier le chevauchement des loci entre les individus. L'incohérence d'une bibliothèque à l'autre peut amplifier les données manquantes. À mesure que la distance évolutive augmente, les sites de restriction partagés peuvent disparaître, et la comparabilité des loci diminue.
Ce ne sont pas des raisons d'éviter le RAD-seq. Ce sont des raisons de le considérer comme une famille de conceptions soigneusement ajustées plutôt que comme un test prêt à l'emploi. En termes pratiques, le GBS l'emporte souvent en matière de dépistage évolutif à grande échelle, tandis que le RAD-seq peut être mieux adapté aux applications où l'architecture des loci, la divergence récente ou la récupération des sites flanquants sont plus importantes.
Dans les projets qui nécessitent ce style de génotypage à représentation réduite, ddRAD-seq peut être un choix plus structuré lorsque un contrôle plus strict sur l'architecture de la bibliothèque est important.
DArTseq : génération efficace de marqueurs dans des systèmes légers en référence
DArTseq occupe un créneau stratégique différent de celui de GBS et RAD-seq. Il repose également sur la réduction de la complexité génomique, mais son attrait principal réside dans son efficacité pratique dans les espèces où l'infrastructure génomique est encore limitée. Dans les systèmes avec des génomes de référence incomplets, des assemblages fragmentés ou des panels de diversité mal caractérisés, les chercheurs ont souvent besoin d'une plateforme capable de générer des marqueurs informatifs à l'échelle du génome avant qu'un écosystème SNP mature ne soit en place.
C'est là que DArTseq devient utile. Sa valeur ne réside pas seulement dans le fait qu'il produit de nombreux marqueurs. Sa valeur est qu'il abaisse la barrière d'entrée pour un large dépistage génomique. Pour les cultures orphelines, les populations de reproduction sous-caractérisées ou les organismes non modèles, cela peut faire la différence entre un projet bloqué et un ensemble de données fonctionnel pour un premier passage. Dans les projets qui ne sont pas encore prêts pour des ressources SNP à l'échelle du génome entièrement standardisées, des voies de découverte larges telles que Génotypage SNP du génome entier ou des flux de travail à représentation réduite peuvent aider à établir la première carte utile de la variation génomique.
Cependant, DArTseq ne doit pas être considéré comme un point de terminaison universel. La génération de marqueurs peut être efficace, mais la conversion des essais en aval n'est pas toujours aussi directe que dans les workflows construits autour de loci SNP explicitement résolus dès le départ. C'est le véritable compromis stratégique. DArTseq peut être très puissant pour le profilage précoce, le dépistage de la diversité et l'analyse comparative large, mais une fois qu'un projet commence à demander une résolution d'intervalle plus précise, une conversion des essais ou une interprétation spécifique aux loci plus standardisée, les chercheurs doivent souvent se diriger vers un format en aval plus étroit.
C'est pourquoi DArTseq est mieux considéré comme un outil de découverte en première ligne dans des systèmes sans référence. Il aide les projets à avancer. Il ne résout pas toujours le dernier kilomètre du déploiement des marqueurs.
Comment choisir le bon système de marquage
Choisir une plateforme de marque devient beaucoup plus facile une fois que les variables d'entrée sont clairement définies. De nombreuses équipes prennent la décision trop tôt. Elles commencent par des noms de plateforme au lieu de la structure du projet. Une meilleure approche consiste à définir d'abord la biologie, ensuite l'architecture des données, et enfin la technologie.
Les variables les plus importantes sont la taille du génome, la ploïdie, la qualité de la référence, le nombre d'échantillons, la densité des marqueurs cibles, la tolérance aux données manquantes et le véritable objectif de l'étude. Cette dernière variable est généralement celle qui évite des erreurs coûteuses. Un projet conçu pour une découverte large ne devrait pas être contraint prématurément à un format de génotypage de routine. Un projet qui nécessite finalement un essai de reproduction répétable ne devrait pas supposer que la plateforme de découverte restera la plateforme finale.
La taille du génome façonne la complexité. Les petits génomes diploïdes avec un contenu de répétition modeste sont beaucoup plus tolérants dans les flux de travail à représentation réduite. Les grands génomes riches en répétitions ne le sont pas. La même stratégie de restriction qui fonctionne bien dans une espèce peut générer une complexité excessive dans une autre. Lorsque l'objectif est de générer une grande matrice de SNP de première passe à travers de nombreux échantillons, Génotypage par séquençage (GBS) peut être un bon choix, mais seulement lorsque la logique enzymatique, la composition génomique et la profondeur de séquençage ont été correctement ajustées.
La ploïdie complique encore la situation. Les systèmes polyploïdes rendent le dosage des allèles et l'appel des génotypes plus difficiles, surtout lorsque la définition des loci est faible ou que la profondeur de lecture est inégale. Dans ces contextes, les chercheurs ont souvent besoin d'une validation plus solide après la découverte. C'est une des raisons pour lesquelles une découverte large peut commencer par ddRAD-seq ou GBS, mais le projet évolue ensuite vers des formats de validation plus étroits une fois que les loci biologiquement utiles sont connus.
La qualité de référence est tout aussi importante. De fortes références améliorent l'alignement des lectures, l'annotation des loci, l'interprétation des SNP et la transférabilité en aval. De faibles références ne rendent pas les projets de marqueurs impossibles, mais elles modifient la logique du choix de la plateforme. Dans les systèmes légers en références, le séquençage à représentation réduite peut être le bon premier pas. Dans les systèmes riches en références, des pipelines de variantes plus larges tels que Appel de variantes devenir beaucoup plus puissant car le projet peut passer plus sereinement des lectures brutes à l'interprétation au niveau des loci.
le nombre d'échantillons modifie l'économie. Un petit projet peut parfois tolérer plus de travail par échantillon si la question biologique est précise. Les grandes études de cohorte récompensent l'évolutivité. C'est pourquoi les chercheurs traitant des centaines d'échantillons se dirigent souvent vers des plateformes qui compressent le coût par échantillon sans sacrifier trop de densité informative. Dans ces situations, Génotypage SNP du génome entier ou les flux de travail de style GBS deviennent souvent attrayants car ils permettent des comparaisons plus larges à travers de grandes populations.
Le besoin en densité de marqueurs est une autre ligne de démarcation. Si l'étude ne nécessite qu'une discrimination modérée entre les accessions, les SSR peuvent encore suffire. Si le projet dépend du resserrement des intervalles, de la couverture de l'LD ou de la logique d'association à l'échelle du génome, des plateformes centrées sur les SNP denses deviennent beaucoup plus appropriées. Une fois cette transition effectuée, le flux de travail s'étend souvent au-delà du seul génotypage et commence à se connecter à l'interprétation au niveau de la population grâce à des services tels que Étude d'Association à l'Échelle du Génome (GWAS).
La tolérance aux données manquantes est la variable que de nombreuses équipes définissent trop tard. Certains projets peuvent absorber une absence modérée de données. D'autres ne le peuvent pas. Si l'objectif est une découverte exploratoire large, certaines données manquantes peuvent être gérables. Si l'objectif est une cartographie précise ou une validation ciblée en aval, la récupération incohérente des loci devient beaucoup plus dommageable. Dans ces cas, le projet bénéficie souvent de passer d'une découverte large à une validation axée sur des régions. Cartographie fine des SNP ou des tests spécifiques à un locus plus étroits.
Un cadre de décision rapide
| Variable de projet | Scénario à pression réduite | Scénario à pression plus élevée | Implication de la sélection par marqueur |
|---|---|---|---|
| Taille du génome / charge en répétitions | Compact, complexité modérée | Grand, riche en répétitions, complexe | Des génomes plus grands et plus répétitifs exigent un contrôle plus strict sur le choix des enzymes, la profondeur et le filtrage. |
| Ploïdie | Diploïde | Polyploïde ou structurellement complexe | Une ploïdie plus complexe augmente la difficulté d'appel et accroît le besoin d'une validation plus rigoureuse. |
| Qualité de référence | Référence solide disponible | Fragmenté ou pas de référence | Des références solides soutiennent une interprétation plus claire des SNP ; des références plus faibles favorisent souvent une découverte à représentation réduite en premier. |
| Nombre d'échantillons | Petit à modéré | Grande cohorte | De grandes cohortes récompensent des plateformes évolutives telles que GBS ou des flux de travail SNP plus larges. |
| Besoin de densité de marqueurs | Modéré | Élevé à très élevé | Les besoins en haute densité orientent le choix vers des séquençages centrés sur les SNP ou des flux de travail basés sur des puces. |
| Tolérance aux données manquantes | Tolérance modérée | Faible tolérance | Une faible tolérance favorise généralement une cohérence de locus plus forte et un suivi plus ciblé. |
| Point de terminaison | Découverte ou enquête générale | Génotypage en routine en aval | La découverte et le déploiement devraient généralement être considérés comme des décisions de plateforme distinctes. |
La règle centrale est simple : la découverte et le déploiement sont des problèmes différents. Une plateforme qui est excellente pour la découverte à l'échelle du génome peut néanmoins ne pas être le bon choix pour le test final validé. De nombreux projets solides utilisent donc un chemin en plusieurs étapes plutôt qu'une seule plateforme permanente.
Systèmes de marqueurs correspondants aux objectifs de recherche
Analyse de la diversité et structure de la population
L'analyse de la diversité dépend de la codominance, de la résolution de la fréquence allélique et d'un nombre suffisant de loci informatifs pour séparer la véritable structure du bruit d'échantillonnage. C'est pourquoi le choix des marqueurs doit commencer par la question analytique, et non par la popularité de la plateforme.
Les SSR continuent de bien performer dans de nombreuses études sur la diversité car ils combinent la codominance avec une forte variabilité par locus. Dans les projets de petite ou moyenne taille, en particulier lorsque des informations multi-alléliques sont précieuses, les SSR restent très pratiques. Cependant, lorsque le projet nécessite une couverture de locus plus large à travers des ensembles d'échantillons plus importants, les flux de travail centrés sur les SNP deviennent plus attrayants. Les chercheurs qui s'orientent dans cette direction commencent souvent par Génotypage SNP du génome entier lorsqu'ils ont besoin de grandes matrices de marqueurs à travers des accès, des populations ou des panneaux de sélection.
Le changement pratique ne consiste pas à passer des "anciens marqueurs" aux "nouveaux marqueurs". Il s'agit de passer d'une haute information par locus à une grande diversité par projet. C'est là la véritable décision.
Phylogénétique et phylogénomique
Le choix des marqueurs phylogénétiques dépend fortement de la profondeur évolutive. Les divergences très récentes bénéficient de fenêtres génomiques denses et localement comparables. Le séquençage à représentation réduite est souvent utile ici car il peut générer de grands ensembles de marqueurs sans le coût d'un resequencement complet.
Le RAD-seq est souvent attrayant dans des contextes de divergence faible à intermédiaire, tels que les radiations récentes, les séparations de populations et la phylogénomique à l'échelle des clades. Son avantage réside dans la largeur avec le contexte de séquence local. Mais cet avantage s'affaiblit à mesure que les taxons deviennent plus divergents. La perte de sites de restriction s'accumule au fil du temps. Une fois que les loci associés aux restrictions homologues ne sont plus partagés de manière fiable entre les lignées, le chevauchement des loci diminue et la comparabilité devient plus difficile à maintenir.
C'est pourquoi les projets axés sur la divergence récente, la structure à l'échelle des clades ou la résolution des complexes d'espèces préfèrent souvent ddRAD-seq sur un flux de découverte plus générique. Un contrôle plus strict de l'architecture des fragments peut être particulièrement précieux lorsque le véritable défi n'est pas simplement de trouver des marqueurs, mais de trouver des marqueurs qui restent comparables entre les taxons exacts analysés.
Cartographie de liaison et cartographie fine
Le mapping modifie à nouveau l'équilibre car la densité affecte directement la résolution. Des marqueurs rares peuvent indiquer qu'une région est importante, mais des marqueurs denses sont nécessaires pour restreindre efficacement cette région. C'est l'un des domaines les plus forts pour GBS et RAD-seq. Les deux peuvent placer de nombreux marqueurs à travers le génome et augmenter la probabilité que des loci informatifs se situent près des points de rupture de recombinaison et des intervalles associés aux traits.
Dans la cartographie en phase initiale, Génotypage par séquençage (GBS) est souvent un choix pratique car il permet une large découverte de marqueurs chez de nombreux individus à un coût gérable. Mais une fois que l'intervalle commence à se réduire, le projet change généralement de caractère. L'ampleur devient moins importante que la confiance régionale. C'est à ce moment-là que Cartographie fine des SNP devient beaucoup plus pertinent, car l'objectif n'est plus de disperser des marqueurs largement à travers le génome. L'objectif est de raffiner l'intervalle biologiquement significatif avec un focus régional plus étroit.
Ce changement scénarisé est l'endroit où de nombreuses pages de ressources restent trop générales. Dans des projets réels, le succès de la cartographie dépend non seulement d'une découverte dense, mais aussi de savoir quand arrêter d'élargir la portée des marqueurs et commencer à intensifier la résolution locale.
GWAS
Les GWAS imposent un ensemble de demandes différent car l'analyse dépend de la structure de déséquilibre de liaison, de la densité des marqueurs, de la composition de la population et de la comparabilité entre les échantillons. Le choix de la plateforme n'est donc pas seulement une question de prix. Il s'agit de savoir si l'ensemble de marqueurs capture la structure de déséquilibre de liaison qui existe réellement dans la population cible.
Si une espèce dispose déjà d'un écosystème SNP mature, des puces ou des panneaux SNP validés peuvent offrir une meilleure comparabilité entre les études. Cela est particulièrement important lorsque la standardisation est cruciale entre les programmes de sélection, les centres de recherche ou les ensembles de données historiques. Cependant, les panneaux établis ne sont pas automatiquement neutres. S'ils ont été construits à partir d'une population de découverte étroite, un biais de constatation peut fausser la représentation dans un germoplasme plus large ou plus diversifié.
GBS offre une alternative plus flexible lorsque les ressources SNP sont rares ou lorsque le projet nécessite encore des découvertes. Mais la flexibilité s'accompagne d'une sensibilité au design. La structure de la bibliothèque, l'absence de données, la profondeur et le filtrage des loci façonnent toutes le jeu de données final. C'est pourquoi le GWAS n'est pas simplement une couche statistique ajoutée après le séquençage. Il fait partie de la décision initiale de la plateforme.
En pratique, de nombreux projets orientés vers les GWAS combinent une approche de génotypage avec un soutien analytique en aval. Un ensemble de données généré par Génotypage SNP du génome entier ou le GBS devient beaucoup plus utile une fois associé à un système robuste Appel de variantes et un défini Étude d'association à l'échelle du génome (GWAS) flux de travail. La valeur ne réside pas seulement dans la génération de marqueurs. Elle se trouve dans la traduction de ces marqueurs en une matrice interprétable et prête pour l'association.
Sélection assistée par marqueurs
La sélection assistée par marqueurs est l'endroit où la différence entre la découverte et le déploiement devient la plus évidente. Un marqueur de découverte doit seulement révéler une variation utile. Un marqueur de sélection doit faire beaucoup plus. Il doit suivre la région pertinente de manière fiable, se comporter de manière cohérente à travers le matériel de reproduction réel, produire de faibles taux d'erreur et rester suffisamment pratique pour une utilisation répétée et routinière.
C'est pourquoi le fort polymorphisme générique est souvent surestimé dans l'élevage. Un marqueur peut être très polymorphe et pourtant être faible pour la sélection s'il est seulement faiblement lié à la véritable cible. Une forte informativité générique ne garantit pas une forte valeur prédictive pour le locus qui compte dans le processus d'élevage.
C'est aussi pourquoi les ensembles de données de découverte dense sont rarement la fin de l'histoire. Un projet peut commencer par Génotypage par séquençage (GBS) ou ddRAD-seq pour identifier des régions candidates. Il peut ensuite passer à Cartographie fine des SNP pour affiner l'intervalle. Une fois que l'ensemble du locus est suffisamment clair, le projet nécessite souvent une voie de validation plus étroite. À ce stade, Services de séquençage d'amplicons peut être utile pour la confirmation de locus ciblé, tandis que Génotypage SNP TaqMan ou Génotypage SNP MassARRAY peut être mieux adapté pour le génotypage ciblé répétable dans un flux de travail de dépistage routinier.
Cette progression est importante car le MAS n'est pas remporté par la plateforme ayant le plus de marqueurs. Il est remporté par la plateforme qui maintient les bons marqueurs stables au moment où de réelles décisions de sélection sont prises.
Une comparaison pratique des principaux systèmes de marqueurs
| Système de marqueurs | Densité typique | Codominance | Reproductibilité | Coût par point de données | Difficulté technique | Applications les mieux adaptées |
|---|---|---|---|---|---|---|
| RFLP | Bas | Oui | Élevé dans les flux de travail qualifiés | Élevé | Élevé | Cartographie historique, travail d'héritage spécifique au lieu |
| AFLP | Faible à modéré | Habituellement dominant | Bon | Modéré | Modéré | Empreinte digitale, dépistage de la diversité, anciens flux de travail non séquentiels |
| SSR | Faible à modéré | Oui | Bon à élevé | Modéré | Modéré | Analyse de la diversité, parenté, structure de la population, cartographie à échelle modérée |
| Panneau SNP / Array SNP | Modéré à très élevé | Oui | Très élevé | Faible une fois établi | Modéré | GWAS, génotypage standardisé, dépistage de grandes cohortes |
| GBS | Élevé à très élevé | Oui | Élevé mais dépendant du design | Bas | Charge bioinformatique modérée à élevée | Découverte de SNP, panneaux de diversité, cartographie de liaison, enquêtes sur de grandes cohortes |
| RAD-seq | Élevé | Oui | Élevé mais sensible au protocole | Bas | Élevé | Cartographie fine, phylogénomique, génomique écologique, espèces non-modèles |
| DArTseq | Élevé | Structure de sortie dépendante de la plateforme | Bon à élevé | Bas | Modéré | Génotypage large dans des systèmes à lumière de référence |
Aucune plateforme ne remporte toutes les catégories. Le RFLP est reproductible mais à faible débit. Les SSR sont très informatifs par locus mais ne sont pas naturellement à l'échelle du génome. Les puces SNP sont standardisées mais dépendent d'écosystèmes de marqueurs préexistants. Le GBS et le RAD-seq sont de puissants moteurs de découverte, mais ils nécessitent une discipline de conception et souvent une conversion ultérieure. Le DArTseq est efficace dans les systèmes légers en référence, mais ce n'est pas toujours le pont le plus clair vers des essais en aval ciblés de manière précise.
C'est pourquoi les tableaux de comparaison ne sont utiles que lorsqu'ils conduisent à des décisions de flux de travail. Une fois qu'un projet doit passer d'une découverte large à une validation plus étroite, les chercheurs cessent souvent de se demander quelle plateforme est "meilleure" et commencent à se demander quelle plateforme est la meilleure. maintenant. Dans cette transition, des outils comme Appel de variantes, Cartographie fine des SNPet les formats de génotypage ciblés deviennent beaucoup plus importants que la seule densité de découverte brute.
Un flux de travail en plusieurs étapes : découverte d'abord, déploiement ensuite.
De nombreux projets de marquage deviennent inefficaces car ils essaient de forcer une seule plateforme à résoudre chaque étape du flux de travail. Une meilleure stratégie est d'adopter une approche par étapes.
Étape 1 : découverte génomique large.
L'objectif ici est de trouver des variations, de décrire la structure ou de localiser des intervalles génomiques. Des approches à haute densité telles que Génotypage par séquençage (GBS), ddRAD-seqou Génotypage SNP du génome entier sont souvent les plus forts à ce stade.
Étape 2 : réduction du locus et validation.
Une fois que les régions informatives sont identifiées, le projet passe de l'étendue à la confiance. La question devient de savoir quels loci restent robustes après filtrage, validation et tests sur des matériaux pertinents. C'est ici que Cartographie fine des SNP et concentré Services de séquençage d'amplicons commencer à jouer un rôle beaucoup plus important.
Étape 3 : génotypage de routine ciblé.
À ce stade, la plateforme gagnante change souvent à nouveau. Un petit nombre de marqueurs performants peut maintenant être déplacé vers des formats d'essai plus propres, plus étroits et plus reproductibles tels que Génotypage SNP TaqMan ou Génotypage SNP MassARRAY.
Cette logique en trois étapes est souvent ce qui sépare un exercice de découverte de marqueurs de niveau recherche d'un flux de travail de marqueurs utilisable. La plateforme de découverte n'a pas besoin d'être permanente. Elle doit simplement fonctionner suffisamment bien pour identifier les bons loci pour la phase suivante.
De l'abondance des marqueurs à une stratégie de marqueur adaptée à l'objectif
Les marqueurs moléculaires d'ADN n'ont pas évolué vers une plateforme parfaite. Ils ont évolué vers différents équilibres de densité, d'informativité, de reproductibilité et de déployabilité.
Les RFLP et AFLP ont établi le principe selon lequel la variation de l'ADN pouvait être suivie de manière systématique. Les SSR ont augmenté la résolution allélique et restent précieux là où des informations multi-alléliques sont importantes. Les systèmes centrés sur les SNP ont transformé le génotypage en un cadre numérique évolutif. Le GBS, le RAD-seq et le DArTseq ont étendu ce cadre en rendant la découverte à haut débit pratique dans des contextes où le resequencement de génome entier serait excessif, inefficace ou trop coûteux.
La stratégie de marqueur la plus utile est donc celle qui correspond à l'objectif final. Pour l'analyse de la diversité, la codominance et les fréquences alléliques interprétables sont les plus importantes. Pour le mapping et les GWAS, l'étendue génomique et la densité des marqueurs deviennent plus importantes. Pour la phylogénomique, la récupérabilité des loci à travers les taxons devient une contrainte déterminante. Pour la sélection, un génotypage en aval robuste est souvent plus important que l'abondance maximale des marqueurs.
Une fois que cette logique est rendue explicite, la sélection des marqueurs devient moins une question de mode des plateformes et davantage une question d'alignement expérimental. Les chercheurs n'ont plus seulement besoin de plus de marqueurs. Ils ont besoin de systèmes de marqueurs qui préservent la bonne information, à la bonne échelle, pour la prochaine étape réelle du flux de travail. En pratique, cela signifie souvent connecter des plateformes orientées vers la découverte avec des services en aval tels que Appel de variantes, Cartographie fine des SNPet des formats de tests ciblés plutôt que de s'attendre à ce qu'une seule plateforme large gère l'ensemble du projet seule.
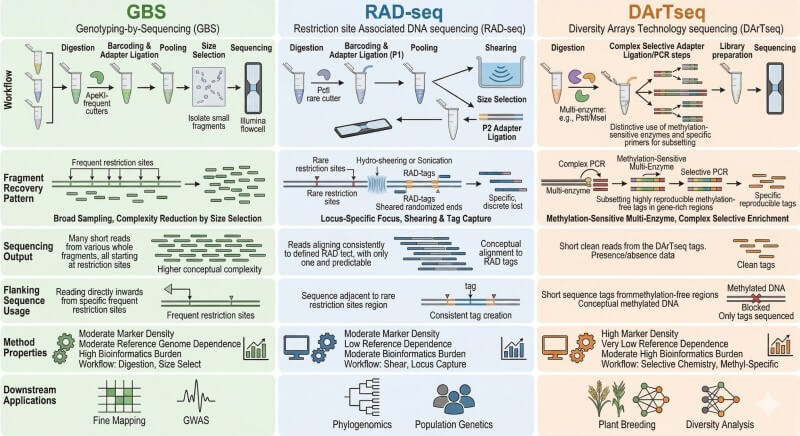 Figure 5. GBS, RAD-seq et DArTseq partagent une logique de représentation réduite, mais diffèrent par l'architecture de récupération des fragments, la transparence des loci et les meilleures applications en aval.
Figure 5. GBS, RAD-seq et DArTseq partagent une logique de représentation réduite, mais diffèrent par l'architecture de récupération des fragments, la transparence des loci et les meilleures applications en aval.
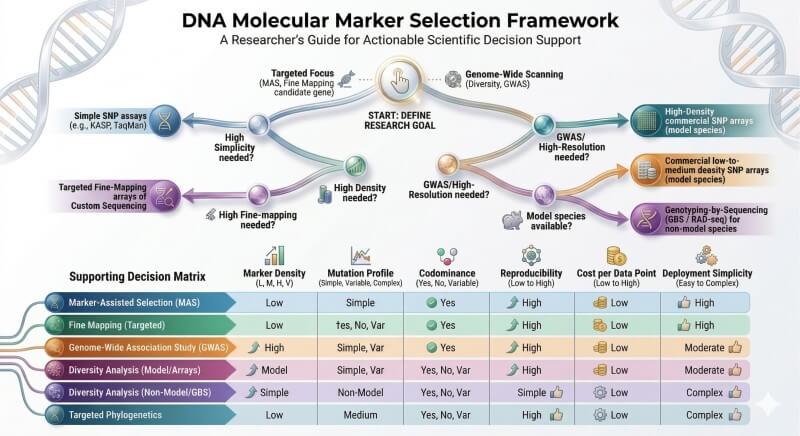 Figure 6. La sélection des marqueurs doit commencer par le point final biologique, puis remonter à travers la complexité du génome, la qualité de référence, les besoins en densité de marqueurs, la tolérance aux données manquantes et le format d'essai en aval.
Figure 6. La sélection des marqueurs doit commencer par le point final biologique, puis remonter à travers la complexité du génome, la qualité de référence, les besoins en densité de marqueurs, la tolérance aux données manquantes et le format d'essai en aval.
FAQ
Quelle est la plus grande différence entre les marqueurs classiques et les marqueurs basés sur le séquençage moderne ?
Les marqueurs classiques s'appuient souvent sur des lectures de motifs de fragments, tandis que les systèmes modernes produisent de plus en plus des génotypes définis par séquence qui sont plus faciles à mettre à l'échelle, à comparer et à intégrer dans une analyse à l'échelle du génome.
Pourquoi les marqueurs codominants sont-ils généralement préférés pour l'analyse de la diversité ?
Parce qu'ils préservent l'intégralité des classes de génotypes et soutiennent des estimations plus robustes de la fréquence allélique, de l'hétérozygotie, de la parenté et de la structure de population.
Quelle quantité de données manquantes est acceptable dans un projet GBS ?
Il n'existe pas de seuil universel. L'absence acceptable dépend de la profondeur de séquençage, de la structure de la population, de la cohérence de la récupération des loci, de la stratégie d'imputation et de l'objectif final du projet. Les enquêtes de diversité exploratoire peuvent tolérer plus d'absences que le cartographie fine ou la validation ciblée en aval.
Quand devrais-je choisir GBS plutôt que le RAD-seq ?
GBS est souvent préféré lorsque la découverte d'SNP évolutifs et économiques à travers de nombreux échantillons est l'objectif principal. Le RAD-seq est souvent plus adapté lorsque l'architecture des loci, la récupération des sites flanquants ou la phylogénomique des divergences récentes sont plus importantes.
DArTseq est-il une bonne option sans génome de référence ?
Oui, souvent. DArTseq peut être particulièrement utile dans des systèmes non-modèles ou à référence limitée où les chercheurs ont besoin de générer un large éventail de marqueurs avant qu'une infrastructure génomique mature n'existe.
Les SSR sont-ils obsolètes maintenant que les méthodes SNP dominent ?
Non. Les SSR continuent de bien fonctionner dans l'analyse de parenté, les études de diversité et les projets où une information multi-allélique élevée par locus est plus précieuse qu'une densité maximale de marqueurs.
Qu'est-ce qui détermine si un marqueur de découverte peut être converti en un test de reproduction de routine ?
Les principaux facteurs sont la qualité de liaison, la spécificité du locus, la reproductibilité, la transférabilité à travers le germoplasme cible et de faibles taux d'erreur dans le flux de travail de dépistage réel.
Pourquoi les marqueurs polymorphes génériques sont-ils parfois faibles pour la sélection assistée par marqueurs ?
Parce que le polymorphisme générique fort ne garantit pas une forte valeur prédictive pour la région du trait cible. Les marqueurs validés spécifiques au trait ont généralement de meilleures performances dans les workflows de sélection que les marqueurs largement informatifs mais faiblement liés.
Références
- Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Exploiter la puissance du RADseq pour la génomique écologique et évolutive. Nature Reviews Genetics. 2016;17(2):81-92. DOI : 10.1038/nrg.2015.28
- Botstein D, White RL, Skolnick M, Davis RW. Construction d'une carte de liaison génétique chez l'homme utilisant des polymorphismes de longueur de fragments de restriction. Journal Américain de Génétique Humaine. 1980;32(3):314-331.
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. Une approche robuste et simple de génotypage par séquençage (GBS) pour des espèces à haute diversité. PLoS ONE. 2011;6(5):e19379. DOI : 10.1371/journal.pone.0019379
- Platten JD, Cobb JN, Zantua RE. Critères d'évaluation des marqueurs moléculaires : Métriques de qualité complètes pour améliorer la sélection assistée par marqueurs. PLoS ONE. 2019;14(1):e0210529. DOI : 10.1371/journal.pone.0210529
- Semagn K, Babu R, Hearne S, Olsen M. Génotypage des polymorphismes nucléotidiques simples par PCR spécifique des allèles compétitifs (KASP) : aperçu de la technologie et de son application dans l'amélioration des cultures. Molecular Breeding. 2014;33(1):1-14. DOI : 10.1007/s11032-013-9917-x
- Serrote CML, Reiniger LRS, Silva KB, dos Santos Rabaiolli SM, Stefanel CM. Détermination du contenu en information de polymorphisme d'un marqueur moléculaire. Gène. 2020;726:144175. DOI : 10.1016/j.gene.2019.144175
- Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, et al. Technologie des Arrays de Diversité : Une technologie de profilage génomique générique sur des plateformes ouvertes. Dans : Méthodes en biologie moléculaire. 2012 ; 888 : 67-89. DOI : 10.1007/978-1-61779-870-2_5
- Li X, Hu Z, Yu W, Xie H, Wang X, Huang P, Zhang X, Yang J, Li Y, Zhao W, et al. Avancées et défis des technologies de marqueurs moléculaires végétaux et de leurs applications à l'ère de l'intelligence artificielle. Frontiers in Plant Science. 2026;16:1757949. DOI : 10.3389/fpls.2025.1757949
- Yadav HK, Solanki RS, Kumar P. Avancées récentes dans la sélection assistée par marqueurs moléculaires et applications dans les programmes de sélection végétale. Journal d'ingénierie génétique et de biotechnologie. 2021;19:128. DOI : 10.1186/s43141-021-00231-1
- Gupta PK, Rustgi S, Kulwal PL. Déséquilibre de liaison et études d'association chez les plantes supérieures : état actuel et perspectives futures. Biologie moléculaire des plantes. 2005;57(4):461-485. DOI : 10.1007/s11103-005-0257-z
À des fins de recherche uniquement, non destiné à un diagnostic clinique, un traitement ou des évaluations de santé individuelles.


