
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Qu'est-ce que le séquençage de nouvelle génération (NGS) ?
Le séquençage de nouvelle génération (NGS) n'est pas une technologie unique. C'est un système de projet qui intègre la préparation des échantillons, la construction de bibliothèques, le choix de la plateforme, les paramètres de séquençage et l'analyse bioinformatique. Le choix de chaque composant détermine si les données finales peuvent répondre à la question biologique d'origine.
Ce guide est conçu pour les chercheurs qui comprennent déjà ce qu'est le séquençage de nouvelle génération (NGS) et qui ont besoin d'un cadre orienté vers la décision pour planifier leur prochain projet de séquençage. Il aborde comment sélectionner la bonne stratégie de séquençage en fonction des objectifs de recherche, des caractéristiques des échantillons, des capacités de la plateforme et des exigences de qualité des données. L'accent est mis sur une logique de conception de projet actionnable, et non sur la répétition des définitions de base du NGS qui sont déjà bien couvertes dans les ressources existantes.
De la technologie de séquençage au système de conception de projet
Le séquençage NGS diffère fondamentalement du séquençage Sanger sur trois aspects qui influencent chaque décision de conception de projet. Premièrement, le NGS lit des millions à des milliards de fragments d'ADN en parallèle, produisant des volumes de données qui nécessitent une infrastructure informatique pour le traitement et l'interprétation. Deuxièmement, le NGS génère des lectures courtes ou longues qui doivent être alignées à une référence ou assemblées de novo—le type de lecture détermine quelles questions biologiques peuvent être abordées. Troisièmement, la qualité des données NGS dépend de la profondeur de séquençage, de l'uniformité de la couverture, des modèles d'erreur et de la qualité de la base de données de référence, et pas seulement de la précision de la plateforme.
Les chercheurs évaluant un projet NGS devraient se concentrer sur cinq questions pratiques avant de choisir une plateforme ou un fournisseur de services :
- Mon type et ma qualité d'échantillon peuvent-ils soutenir l'analyse prévue ?
- Devrais-je choisir une stratégie de séquençage à courtes lectures, à longues lectures ou hybride ?
- Quelle profondeur de séquençage ai-je besoin pour des résultats fiables ?
- Quels indicateurs de qualité devrais-je suivre à chaque étape ?
- Quelles questions biologiques les données résultantes peuvent-elles répondre, et quelles questions restent en dehors de son champ d'application ?
Ces questions constituent l'épine dorsale du cadre de conception de projet décrit dans ce guide. Pour les chercheurs qui commencent leur premier projet NGS ou évaluent une nouvelle application, services NGS complets fournir des conseils d'expert sur la conception expérimentale et la sélection de stratégies.
 Figure 1. Pile de conception de projet NGS — de l'échantillon à l'interprétation biologique
Figure 1. Pile de conception de projet NGS — de l'échantillon à l'interprétation biologique
Légende : Pile de conception de projet NGS à cinq couches montrant la progression de la question biologique à la préparation des échantillons, la construction de la bibliothèque, le choix de la plateforme, les paramètres de séquençage et l'analyse bioinformatique jusqu'à l'interprétation biologique.
De la question de recherche à la stratégie NGS — Un cadre décisionnel
L'erreur la plus courante dans la planification de projets NGS est de commencer par la plateforme plutôt que par la question biologique. La bonne stratégie de séquençage dépend de ce que la recherche vise à découvrir, mesurer ou comparer.
Classification des objectifs de recherche par exigence NGS : La première étape dans la conception d'un projet NGS est de classer l'objectif de recherche en fonction du type d'information biologique requise. Chaque type d'objectif a des exigences spécifiques en matière de paramètres de séquençage qui déterminent le choix de la plateforme, de la profondeur et de l'analyse des données.
| Objectif de recherche | Meilleure stratégie NGS | Variable de conception clé | Risque en cas de désalignement |
|---|---|---|---|
| Détection de SNP / petites InDel | Séquençage génomique à lecture courte / Séquençage de l'exome entier / panneau ciblé | Profondeur, qualité de cartographie, taux de duplication | Appels à faible confiance |
| Découverte de variants structuraux | Séquençage génomique à lecture longue ou hybride | Longueur de lecture, intégrité de la molécule | SVs manqués |
| Assemblage de génome de novo | PacBio HiFi / ONT / hybride | N50, couverture, hétérozygotie | Assemblage fragmenté |
| Expression différentielle | RNA-seq | Intégrité de l'ARN, type de bibliothèque, réplicats biologiques | Interprétation biologique erronée |
| Profilage du microbiome | Métagénomique par amplicon ou par shotgun | Région marqueur, base de données, épuisement de l'hôte | Profil taxonomique biaisé |
Logique de décision : Si un génome de référence de haute qualité est disponible pour l'espèce cible, le séquençage à lecture courte est l'approche la plus rentable pour la détection et la quantification des variants dans la plupart des applications. Le compromis est une résolution limitée dans les régions répétées. Si la recherche nécessite de résoudre des régions répétées plus longues que la longueur de lecture, de détecter de grands variants structurels (délétions, insertions, inversions dépassant 50 pb) ou d'assembler un génome de novo, le séquençage à lecture longue est nécessaire malgré son coût par base plus élevé et des exigences d'échantillon plus strictes.
Pour les projets nécessitant à la fois précision et continuité, des stratégies hybrides combinant des plateformes de lectures courtes et longues offrent le meilleur équilibre. Une assemblage génomique hybride typique utilise des lectures longues PacBio HiFi ou Nanopore pour la construction de contigs et des lectures courtes Illumina pour le polissage et la correction des erreurs. Le coût de cette approche à double plateforme est plus élevé, mais la qualité d'assemblage résultante justifie l'investissement pour les projets de haute priorité.
Erreurs courantes à éviter :
- Demander "combien de gigabases" sans d'abord définir la question biologique.
- Comparer les prix des plateformes sans évaluer si les données résultantes peuvent soutenir la conclusion prévue.
- Traiter le séquençage WGS, WES, RNA-seq et le séquençage d'amplicons comme des types de projets interchangeables.
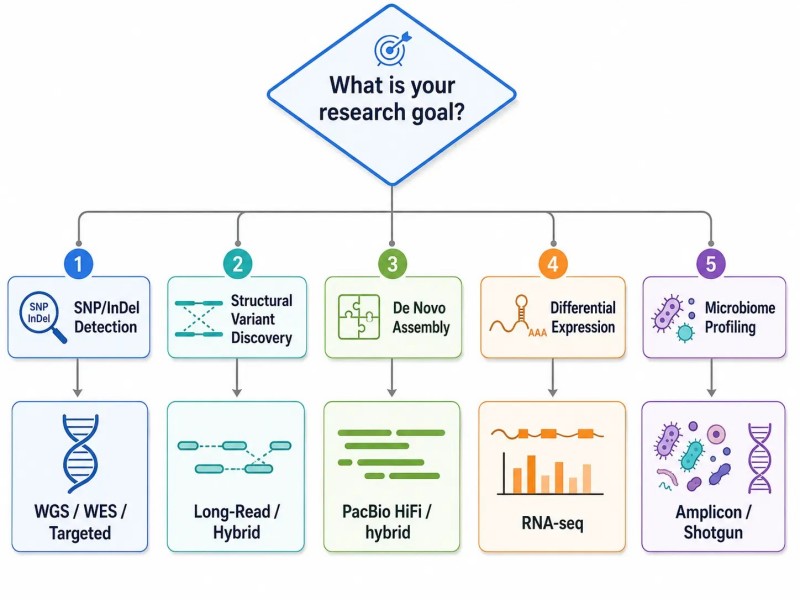 Figure 2. Arbre de décision pour la stratégie NGS en fonction de la question de recherche
Figure 2. Arbre de décision pour la stratégie NGS en fonction de la question de recherche
Légende : Carte d'arbre de décision reliant cinq objectifs de recherche courants—détection de SNP, découverte de variants structurels, assemblage de novo, expression différentielle et profilage du microbiome—à leurs stratégies NGS optimales avec des variables de conception clés et des risques de désalignement.
Sélection de la plateforme — Comment évaluer l'adéquation de la plateforme pour votre projet
La sélection de la plateforme ne consiste pas à classer les technologies, mais à faire correspondre leurs caractéristiques aux exigences spécifiques d'un projet de recherche. L'étude sur le séquençage de nouvelle génération de l'ABRF a démontré que différentes plateformes produisent des résultats mesurablement différents en termes de cohérence de couverture, de taux d'erreur et de performance de détection des variants. Ces différences signifient que le choix de la plateforme affecte directement les découvertes biologiques qui peuvent être réalisées.
Considérations clés au niveau du projet : Pour les projets où le débit et la précision par base sont les principales exigences—détection de SNV, quantification RNA-seq, séquençage d'exome et panels ciblés—le séquençage à courtes lectures est l'approche la plus établie. services NGS Le portefeuille comprend plusieurs plateformes de séquençage à lecture courte pour adapter le débit à l'échelle du projet.
Pour les projets nécessitant la résolution de régions génomiques plus longues que la longueur de lecture—assemblage de novo, détection de variants structurels et séquençage de transcrits complets—des plateformes à longues lectures sont nécessaires malgré un coût par base plus élevé et des exigences d'échantillonnage plus strictes. Un détail comparaison des technologies PacBio et Oxford Nanopore est disponible pour les chercheurs évaluant les options de lecture longue.
Pour les projets nécessitant à la fois précision et continuité—assemblage génomique complet ou détection complète de variantes—les stratégies hybrides combinant le séquençage à lecture courte et à lecture longue offrent le meilleur équilibre. Cette approche à double plateforme nécessite un investissement total plus élevé mais produit une qualité de données qu'aucune des plateformes ne peut atteindre seule.
Erreurs courantes dans le choix de la plateforme : Supposer que des lectures plus longues sont universellement meilleures, supposer que des lectures courtes ne peuvent pas contribuer à l'analyse des variants structurels, et s'attendre à ce qu'une seule plateforme soit optimale pour tous les types de projets figurent parmi les erreurs les plus fréquentes dans la conception de projets NGS.
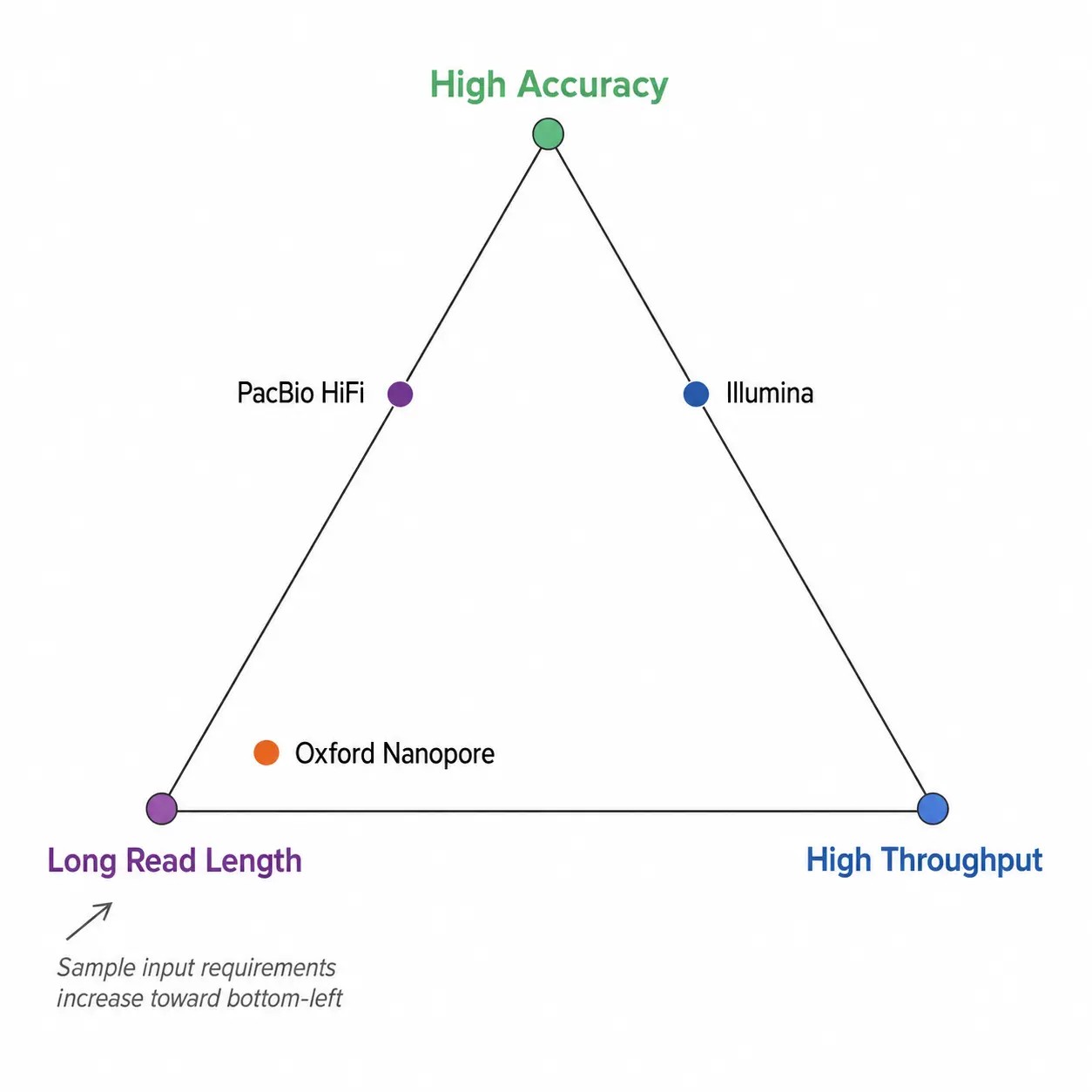 Figure 3. Triangle de compromis des plateformes NGS — précision, longueur de lecture, débit et exigences en matière d'échantillons
Figure 3. Triangle de compromis des plateformes NGS — précision, longueur de lecture, débit et exigences en matière d'échantillons
Triangle de compromis illustrant l'interrelation entre la précision, la longueur de lecture, le débit et les exigences d'échantillonnage à travers les plateformes NGS à lecture courte, à lecture longue et hybrides.
La qualité de l'échantillon est la première contrainte dans tout projet de NGS.
Aucune profondeur de séquençage ou bioinformatique sophistiquée ne peut compenser une mauvaise qualité d'échantillon. L'évaluation de la qualité de l'échantillon devrait être la première étape dans la conception du projet, avant le choix de la plateforme ou de la bibliothèque.
Variables clés pour les échantillons d'ADN : La quantité d'entrée (masse totale et concentration), la distribution de la taille des fragments, le niveau de dégradation (évalué par électrophorèse sur gel ou TapeStation), les rapports de pureté (A260/280, A260/230) et les contaminants (phénol, éthanol, sels, polysaccharides, hème, acide humique) affectent tous l'efficacité de la construction de la bibliothèque et la qualité des données de séquençage. Un échantillon qui réussit le contrôle de qualité de concentration mais contient du phénol résiduel échouera à l'étape de ligation car le phénol inhibe l'ADN ligase. Pour cette raison, l'évaluation de la pureté par spectrophotométrie combinée à la quantification fluorométrique fournit une image de qualité plus fiable que l'une ou l'autre méthode seule.
Seuils de contrôle qualité quantitatifs pour l'ADN : A260/280 devrait être compris entre 1,8 et 2,0 pour de l'ADN pur ; des valeurs en dehors de cette plage indiquent une contamination par des protéines ou du phénol. A260/230 devrait être compris entre 2,0 et 2,2 ; des valeurs plus basses suggèrent un résidu de composé organique ou de glucides. Pour l'ADN de haut poids moléculaire requis par les plateformes de séquençage à longues lectures, l'ADN génomique devrait montrer une bande dominante au-dessus de 20 kb sur un gel ou un trace TapeStation sans bavure significative en dessous de 10 kb.
Variables clés pour les échantillons d'ARN : Score RIN (RIN ≥ 7 pour mRNA-seq, RIN ≥ 5 pour total RNA-seq), DV200 pour les échantillons d'ARN FFPE, niveau de contamination par l'ARNr et méthode de préservation des tissus. L'ARN dérivé de FFPE nécessite des protocoles de préparation de bibliothèque spécifiques avec des étapes de réparation des dommages, et le rendement attendu est généralement inférieur à celui des tissus frais congelés.
Considérations supplémentaires pour le séquençage à longues lectures : L'extraction d'ADN à haut poids moléculaire est essentielle. Un maniement délicat lors de l'extraction, un minimum de cycles de congélation-dégel et l'évitement de la cisaillement mécanique lors de la pipetage sont cruciaux pour préserver les longs fragments nécessaires à la préparation de bibliothèques PacBio et Nanopore.
Diagnostiquer les problèmes de qualité courants :
- Couverture inégale : Biais GC, biais de fragmentation ou faible complexité de la bibliothèque. Solution : réévaluer la qualité de l'entrée, ajuster la méthode de bibliothèque, contrôler les cycles de PCR.
- Taux de cartographie faible : Contamination, inadéquation du génome de référence ou dégradation de l'échantillon. Solution : ajouter un dépistage de contamination, vérifier l'adéquation du référentiel.
- Taux de duplication élevé : Faible quantité d'ADN, amplification PCR excessive ou faible complexité de bibliothèque. Solution : réduire le nombre de cycles PCR, optimiser la complexité de la bibliothèque, envisager des protocoles sans PCR. Pour les types d'échantillons avec un matériel d'entrée limité, analyse des données génomiques peut aider à évaluer si le taux de duplication est dans des limites acceptables pour la méthode de détection prévue.
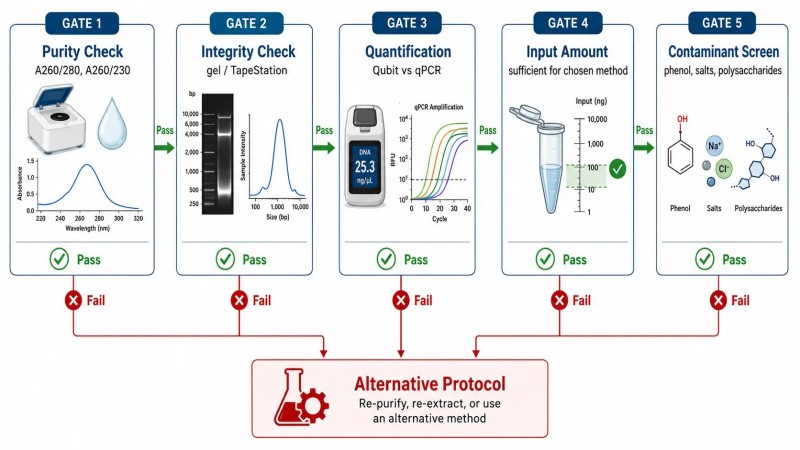 Figure 4. Portes de qualité d'échantillon avant NGS — Seuils de CQ et évaluation des risques
Figure 4. Portes de qualité d'échantillon avant NGS — Seuils de CQ et évaluation des risques
Légende : Exemples de contrôles de qualité pour les échantillons d'ADN et d'ARN avant le séquençage de nouvelle génération (NGS), montrant les seuils de contrôle qualité quantitatifs (A260/280, A260/230, RIN, DV200) et l'évaluation des risques pour les problèmes de qualité courants, y compris la couverture inégale, le faible taux de mappage et le taux de duplication élevé.
La préparation de la bibliothèque détermine l'utilisabilité des données.
La préparation de la bibliothèque est le lien entre l'acide nucléique brut et les molécules prêtes pour le séquençage. Ses fonctions principales sont de convertir l'ADN ou l'ARN en molécules compatibles avec la plateforme, d'introduire des adaptateurs et des codes-barres pour la liaison à la cellule de flux et l'identification des échantillons, de contrôler la taille des inserts et la complexité de la bibliothèque, et de préserver l'information sur les brins lorsque cela est nécessaire.
Variables clés qui affectent la sortie de séquençage : La stratégie de fragmentation (mécanique vs. enzymatique vs. tagmentation) affecte le biais de couverture et la reproductibilité. L'efficacité de la ligation des adaptateurs détermine la proportion de fragments pouvant être séquencés. Le nombre de cycles de PCR influence directement le taux de duplication : chaque cycle supplémentaire au-delà de 10 ajoute environ 5 à 10 % de duplicats supplémentaires. La fenêtre de sélection de taille contrôle la distribution de la taille des inserts, ce qui affecte la densité des clusters et les taux de cartographie. La méthode de quantification de la bibliothèque (qPCR vs. Qubit vs. Bioanalyzer) doit être choisie avec soin : la qPCR est la plus précise pour déterminer la concentration prête pour le séquençage.
Erreurs courantes lors de la préparation de bibliothèques :
- En supposant qu'une forte concentration de bibliothèque équivaut à une haute qualité de bibliothèque.
- En ignorant la contamination par les dimères d'adaptateurs, ce qui gaspille des lectures de séquençage.
- Utiliser la quantification totale de l'ADN (Qubit) seule au lieu de mesurer également les molécules amplifiables (qPCR)
- Amplification PCR excessive dans des échantillons à faible entrée, entraînant des taux de duplication élevés.
Pour une discussion détaillée sur l'optimisation de la préparation des bibliothèques, voir le Ressource de préparation de bibliothèque NGS, qui couvre en profondeur la fragmentation, la réparation des extrémités, la ligation des adaptateurs, l'amplification, le nettoyage et le contrôle de qualité. Pour les projets impliquant des types d'échantillons spécialisés tels que les tissus FFPE ou l'ADNcf, approches de séquençage ciblé nécessitent souvent des protocoles de bibliothèque spécifiques optimisés pour des matériaux dégradés ou à faible entrée.
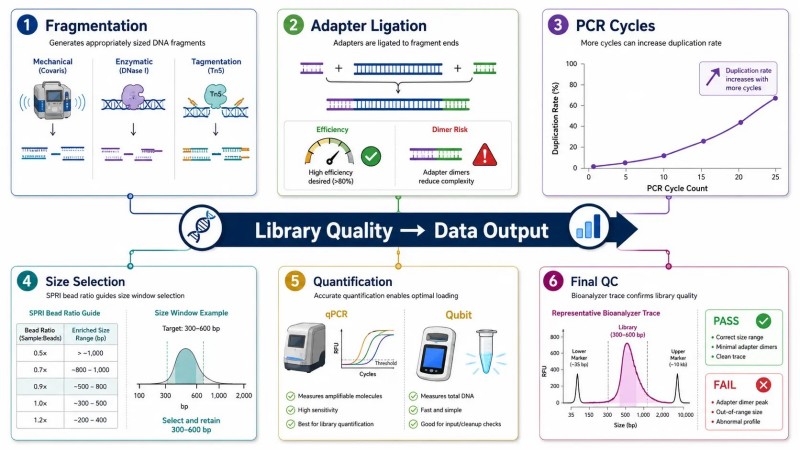 Figure 5. Variables de préparation de bibliothèque qui affectent le rendement du séquençage
Figure 5. Variables de préparation de bibliothèque qui affectent le rendement du séquençage
Légende : Principales variables de préparation de bibliothèque affectant la sortie de séquençage NGS — stratégie de fragmentation, efficacité de ligation des adaptateurs, nombre de cycles PCR, fenêtre de sélection de taille et méthode de quantification — avec erreurs courantes et leur impact sur la qualité des données.
Longueur, profondeur et couverture — trois concepts distincts
Ces trois termes sont souvent utilisés de manière interchangeable dans les discussions de projet, mais ils décrivent différents paramètres qui affectent chacun la qualité des données et le coût du projet de manière indépendante. Le NGS peut être appliqué à l'ADN ou à l'ARN provenant de pratiquement toute source biologique : sang, tissu, cellules, blocs FFPE, plasma (cfDNA), cultures microbiennes, échantillons environnementaux et cellules uniques. La contrainte clé est que la qualité de l'échantillon doit répondre aux exigences de la méthode de préparation de bibliothèque et de la plateforme de séquençage choisies.
Longueur de lecture Le nombre de bases contiguës est déterminé par lecture de séquençage. Cela affecte la précision de l'alignement, la capacité à couvrir des régions répétitives, la résolution des isoformes en RNA-seq et la continuité de l'assemblage. Des lectures plus longues ne sont pas toujours meilleures : elles nécessitent des temps de course plus longs et produisent moins de lectures totales par cellule de flux.
Profondeur de séquençage (le taux de couverture) est le nombre moyen de fois que chaque base dans la région cible est séquencée. Il détermine la confiance dans les appels de variants : une profondeur plus élevée permet de détecter des variants de fréquence plus faible et offre une puissance statistique plus robuste pour l'analyse de l'expression différentielle.
Couverture peut faire référence soit à la fraction du génome cible qui est couverte par au moins une lecture (ampleur de la couverture), soit à la distribution de la profondeur à travers le génome (uniformité). Les discussions de projet devraient préciser quelle signification est intended.
| Métrique | Ce que cela mesure | Pourquoi c'est important | Interprétation erronée courante |
|---|---|---|---|
| Longueur de lecture | Longueur des lectures de séquençage | Alignement, assemblage, répétition de la portée | Plus long signifie toujours mieux. |
| Données brutes | Sortie totale avant filtrage | Échelle de course | Données utilisables |
| Données propres | Lectures de haute qualité filtrées | Entrée en aval | Garantit la qualité de la cartographie |
| Profondeur | Lectures moyennes par locus | Confiance dans la variante | Identique dans toutes les régions du génome |
| Uniformité de couverture | Distribution de la profondeur | Fiabilité à travers les régions | Ignoré si la profondeur moyenne semble élevée. |
Exigences de profondeur spécifiques au projet : La détection des SNV germinaux par séquençage du génome humain nécessite une couverture de 30× comme référence standard. La détection des mutations somatiques dans le cancer nécessite une couverture de 60 à 100× pour identifier des variantes à faible fréquence. L'analyse de l'expression génique par RNA-seq nécessite entre 20 et 50 millions de lectures par échantillon. Le profilage des amplicons 16S nécessite entre 10 000 et 50 000 lectures par échantillon. Ces cibles doivent être utilisées comme valeurs minimales, avec un ajout de 10 à 20 % de sur-séquençage pour tenir compte de la variation de qualité spécifique à chaque échantillon.
Interprétation spécifique au projet : Pour le séquençage génomique complet (WGS), l'accent doit être mis sur la profondeur et l'uniformité à l'échelle du génome. Un séquençage WGS à 30× où certaines régions sont couvertes à 5× et d'autres à 60× n'est pas équivalent à un séquençage où toutes les régions sont couvertes à 25-35×. Les métriques d'uniformité de couverture telles que le coefficient de variation (CV) de la profondeur à travers les bins ou la fraction du génome dans une plage de 0,2× à 2× de la profondeur moyenne fournissent une image de qualité plus complète que la profondeur moyenne seule. Pour le séquençage d'exomes (WES) et les panels ciblés, les métriques clés sont le taux de ciblage, la couverture cible et l'uniformité de capture — et non le total de données produites. Pour le RNA-seq, le nombre de lectures mappées par échantillon, la couverture des corps de gènes et la polarité des bibliothèques sont plus informatifs que le simple nombre de lectures brutes. Pour la métagénomique, la proportion de lectures hôtes, la récupération de la diversité microbienne et les seuils de détection des taxons rares déterminent si la profondeur est adéquate.
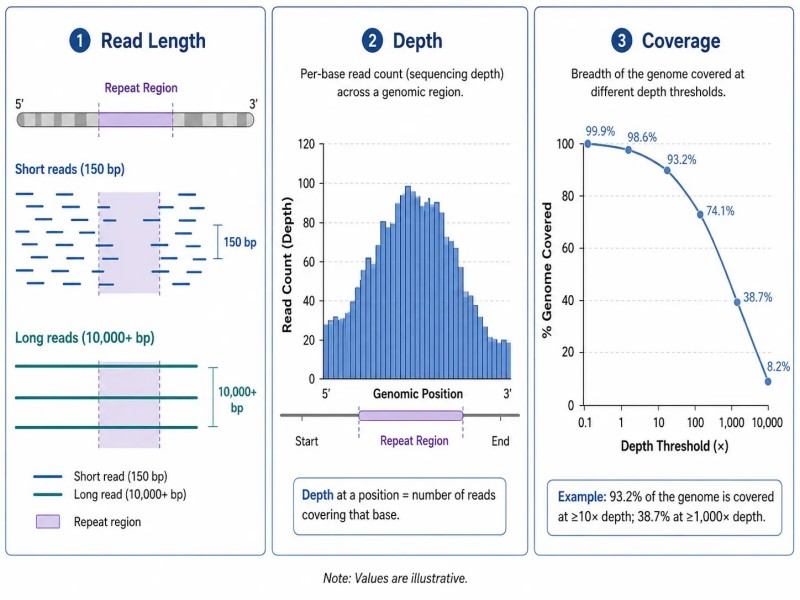 Figure 6. Longueur de lecture vs. profondeur vs. couverture — trois métriques indépendantes dans la conception de projets NGS
Figure 6. Longueur de lecture vs. profondeur vs. couverture — trois métriques indépendantes dans la conception de projets NGS
Légende : Diagramme conceptuel distinguant la longueur de lecture, la profondeur de séquençage et la couverture comme trois métriques NGS indépendantes, avec un tableau clarifiant les définitions, l'importance pratique et les interprétations erronées courantes pour chaque métrique.
Métriques de qualité des données NGS — Que rechercher dans un rapport de séquençage
Un rapport QC complet devrait inclure des métriques à trois niveaux : la qualité au niveau du séquençage, la qualité au niveau de l'alignement et la qualité au niveau de la bibliothèque. Les chercheurs évaluant un fournisseur de services de séquençage devraient savoir quelles métriques sont standard et comment les interpréter.
Contrôle de qualité de séquençage : Pour un run Illumina standard, plus de 85 % des bases devraient être au-dessus de Q30 pour des runs de 2 × 150 pb. La carte thermique de qualité par cycle devrait montrer un déclin progressif de la haute à la qualité modérée — une chute brutale à un numéro de cycle indique un problème spécifique au run qui devrait être examiné avant de procéder à l'analyse des données.
Contrôle qualité de l'alignement : Taux de cartographie, pourcentage de lectures correctement appariées, distribution de la taille des insertions, profondeur de couverture moyenne et uniformité de la couverture. Les faibles taux de cartographie (<80 % pour l'ADN humain) doivent être examinés — les causes possibles incluent la contamination (ADN bactérien, fongique ou humain provenant de la manipulation), l'inadéquation du génome de référence (mauvaise espèce ou version du génome) ou la dégradation de l'échantillon produisant des fragments qui ne peuvent pas s'aligner de manière unique.
Contrôle de qualité de la bibliothèque : Concentration de la bibliothèque, molarité, distribution de taille, contenu en dimères d'adaptateurs et estimation de la complexité de la bibliothèque. Un contenu en dimères d'adaptateurs supérieur à 5 % de la masse totale de la bibliothèque gaspille la capacité de séquençage.
Contrôle qualité spécifique au projet : Pour les WES ou les panels ciblés, le taux d'atteinte et la couverture cible à des profondeurs spécifiées (par exemple, % des bases cibles à 20×, 50×, 100×) sont essentiels. Services de séquençage de l'exome entier Ces métriques sont généralement rapportées comme des livrables standard. Pour le RNA-seq, le taux d'ARNr, la distribution exonique/intronique/intergènique et la couverture des gènes doivent être rapportés. Pour la métagénomique, le niveau de contamination par l'hôte, le taux d'attribution taxonomique et la version de la base de données doivent être documentés. Pour le séquençage à longues lectures, le N50 des lectures, la distribution des longueurs de lecture, le rendement total et l'exactitude brute par rapport à l'exactitude corrigée sont des métriques clés.
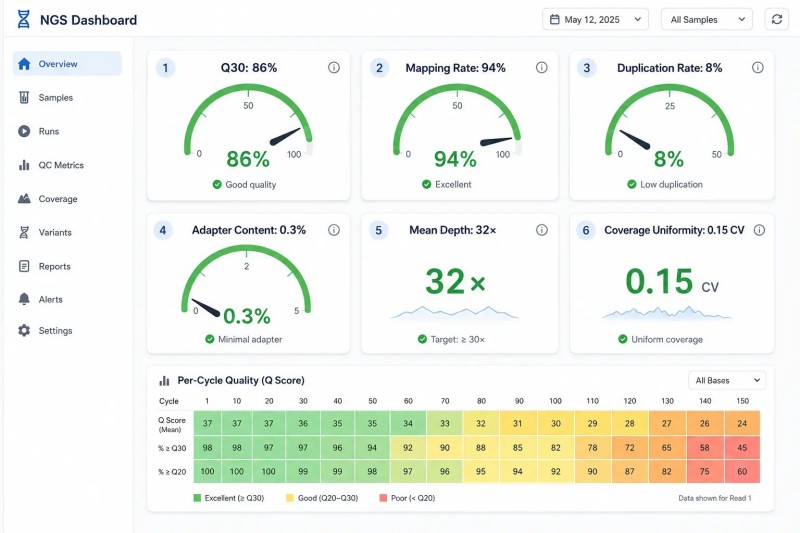 Figure 7. Tableau de bord QC NGS — rapport d'échantillon montrant les indicateurs de qualité clés
Figure 7. Tableau de bord QC NGS — rapport d'échantillon montrant les indicateurs de qualité clés
Légende : Tableau de bord QC NGS complet montrant trois niveaux de métriques de qualité : QC de séquençage (Q30, contenu en GC, contenu en adaptateurs), QC d'alignement (taux de mappage, taille d'insertion, uniformité de couverture) et QC de bibliothèque (concentration, contenu en dimères) - avec des indicateurs spécifiques au projet pour WES, RNA-seq, métagénomique et données de lecture longue.
Analyse bioinformatique — La valeur du NGS réside dans la question, pas dans le séquenceur.
L'instrument de séquençage produit des données brutes. L'analyse bioinformatique transforme ces données en informations biologiques. Le choix de la chaîne d'analyse doit être déterminé par la question de recherche, et non par des paramètres par défaut ou des flux de travail standard.
Composants de la chaîne d'analyse des noyaux : Contrôle qualité des données brutes, découpage et filtrage, alignement ou assemblage, quantification ou appel de variants, annotation, analyse statistique et interprétation biologique. Chaque étape présente des variations spécifiques à la plateforme et à l'application qui influencent les résultats.
Différences clés d'analyse par type de projet :
| Projet NGS | Sortie de bioinformatique fondamentale | Dépendance clé | Question BOFU |
|---|---|---|---|
| WGS | Liste des variantes, annotation, SV/CNV | Qualité de référence, profondeur | Ce design peut-il détecter le type de variante de ma cible ? |
| RNA-seq | DEG, voie, profil d'expression | Qualité de l'ARN, répliques | Le design est-il statistiquement interprétable ? |
| Métagénomique | Taxonomie, fonction, diversité | Base de données, épuisement de l'hôte | Des taxons rares ou des gènes fonctionnels peuvent-ils être résolus ? |
| Assemblage de longue lecture | Contigs, N50, BUSCO, annotation | ADN HMW, couverture | La continuité de l'assemblage est-elle suffisante pour l'objectif de recherche ? |
Erreurs courantes en bioinformatique : En supposant que le même pipeline fonctionne pour tous les types de projets, en ignorant la qualité et la version du génome de référence, en négligeant l'impact de la version de la base de données sur les résultats d'annotation, en concevant des expériences sans réplicats biologiques ou modèles statistiques appropriés, et en confondant la visualisation des données avec une conclusion biologique. Pour les projets qui nécessitent une personnalisation. analyse bioinformatiqueDiscuter des options de pipeline avec le fournisseur de services avant le début du séquençage garantit que le format de sortie des données correspond aux exigences d'analyse.
Services d'analyse en bioinformatique peut fournir des pipelines adaptés à des types de projets spécifiques, garantissant que le traitement et l'interprétation des données s'alignent sur les objectifs de recherche.
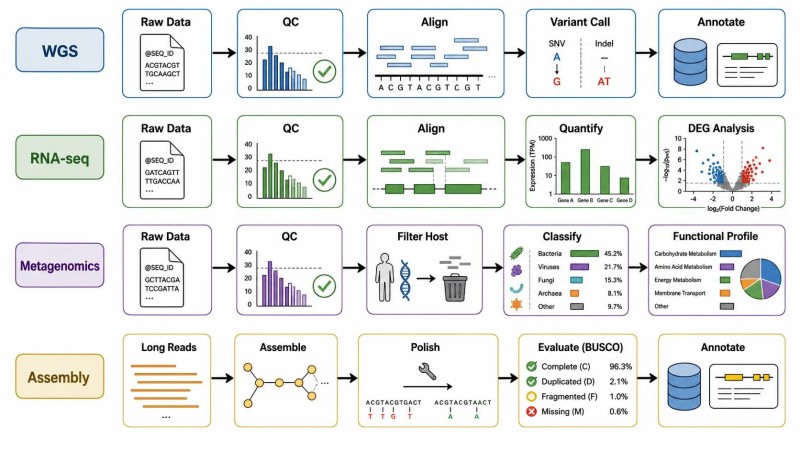 Figure 8. Flux de travail en bioinformatique par type de projet NGS
Figure 8. Flux de travail en bioinformatique par type de projet NGS
Légende : Flux de travail d'analyse bioinformatique pour quatre types de projets NGS — WGS, RNA-seq, métagénomique et assemblage de longues lectures — montrant les composants clés du pipeline, les dépendances essentielles et la question BOFU (Base d'Utilisation Future) qui doit être répondue avant la sélection du pipeline.
Conception d'applications NGS — WGS, WES, RNA-seq, Métagénomique, et plus encore
Séquençage du génome entier
Le séquençage génomique complet (WGS) est approprié pour la découverte de variants à l'échelle du génome, la génomique des populations, l'assemblage de novo et la génomique comparative. Les principales variables de conception incluent la taille du génome, le taux d'hétérozygotie, le contenu en répétitions, le type de variant requis et la disponibilité du génome de référence. Le WGS à lecture courte à 30× est standard pour la détection des SNV germinaux humains. Le WGS à lecture longue est préféré pour l'assemblage, la détection de variants structurels et les régions riches en répétitions. Les stratégies hybrides combinant les deux types de lectures offrent le meilleur équilibre pour une analyse génomique complète.
Pour un projet de séquençage génomique humain, le livrable standard comprend environ 90-100 Go de données brutes par échantillon à 30×. Le pipeline bioinformatique doit gérer l'appel de variants pour les SNVs, les petites indels et les variants de nombre de copies comme minimum, avec une analyse des variants structurels en option. Services de séquençage de génome complet peut être configuré pour des approches à lecture courte, à lecture longue ou hybrides en fonction des objectifs de recherche.
Séquençage de l'exome entier / Séquençage ciblé
Les WES et les panels ciblés se concentrent sur des régions génomiques spécifiques, réduisant les coûts tout en permettant une profondeur plus élevée sur les régions cibles. Les variables clés de conception incluent la conception de la région de capture, les attentes en matière de taux de ciblage, les exigences de couverture des cibles, la compatibilité des sondes avec l'espèce cible et les régions cibles riches en GC ou répétitives. Les risques incluent une couverture inégale des régions cibles, un biais de capture et l'incapacité à interpréter les régions non ciblées.
Pour le séquençage de l'exome humain, un livrable typique inclut une profondeur moyenne de 100 à 200× sur les régions cibles, avec au moins 90 % des bases cibles couvertes à 20× ou plus. Le taux de ciblage (pourcentage de lectures se chevauchant dans ou près du design de capture) doit dépasser 60 % pour les kits de capture d'exome standard.
Séquençage de l'ARN
L'RNA-seq mesure l'expression génique, détecte l'épissage alternatif, identifie les transcrits de fusion et découvre des transcrits nouveaux. Les variables clés incluent l'intégrité de l'ARN, la directionnalité de la bibliothèque (préservant l'orientation des brins), la sélection de poly(A) par rapport à la stratégie de déplétion de l'ARNr, et le nombre de répliques biologiques. Les risques incluent la dégradation de l'ARN affectant la précision de la quantification, les effets de lot dus au traitement des échantillons à des moments différents, et un design de réplique insuffisant pour la puissance statistique.
Un projet standard de séquençage d'ARNm nécessite 20 à 50 millions de lectures par échantillon pour la quantification au niveau des gènes. Pour une analyse au niveau des isoformes, plus de 100 millions de lectures par échantillon peuvent être nécessaires. Au moins trois répliques biologiques par condition sont recommandées pour une analyse d'expression différentielle fiable. services de séquençage d'ARN soutenir à la fois les types de bibliothèques sélectionnées par poly(A) et celles dépourvues d'ARNr.
Séquençage métagénomique
Les métagénomiques profilent la structure de la communauté microbienne, le potentiel fonctionnel et la composition au niveau des souches. Les variables clés incluent la proportion d'ADN hôte, la biomasse microbienne, le choix de la base de données et la profondeur de séquençage. Les risques comprennent la contamination par l'hôte qui peut submerger les lectures microbiennes, une profondeur insuffisante pour la détection des taxons rares, et un biais d'annotation dépendant de la base de données qui varie en fonction de l'ensemble de référence utilisé.
Pour la métagénomique par shotgun, 50 à 100 millions de lectures par échantillon sont typiques pour un profilage fonctionnel complet. Pour le séquençage d'amplicons 16S, 10 000 à 50 000 lectures par échantillon suffisent pour l'analyse de la composition de la communauté. Des stratégies de déplétion de l'ADN hôte — y compris la lyse différentielle et la capture par sondes — doivent être envisagées pour les échantillons de microbiome à faible biomasse provenant de sites associés à l'hôte. Services de séquençage d'amplicons 16S/ITS fournir des protocoles standardisés pour le profilage communautaire, tout en services de métagénomique par shotgun offrir une résolution plus élevée pour l'analyse fonctionnelle et au niveau des contraintes.
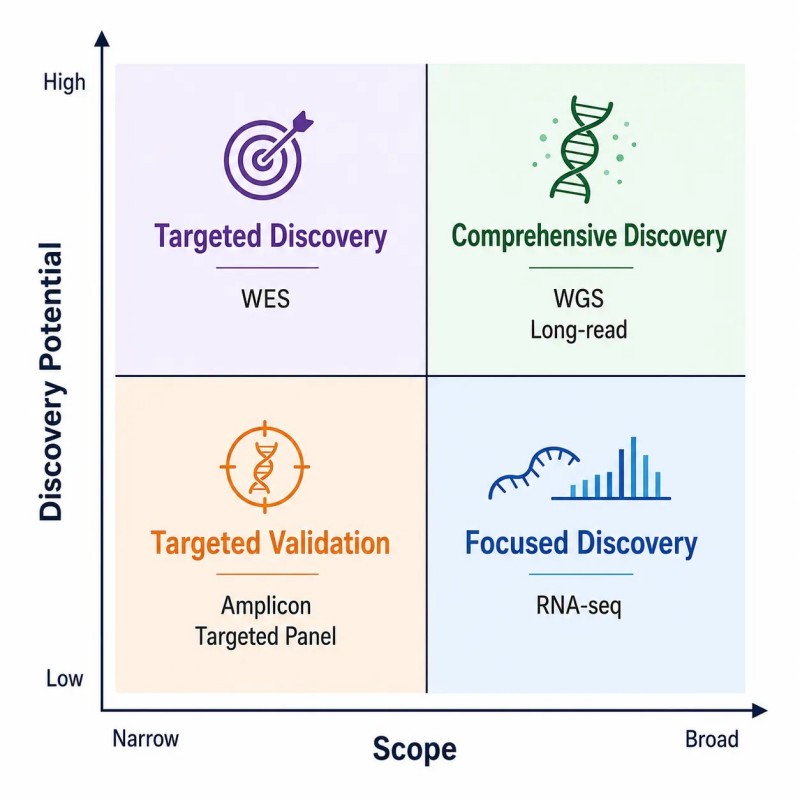 Figure 9. Matrice de sélection des applications NGS — correspondance des objectifs de recherche aux services de séquençage
Figure 9. Matrice de sélection des applications NGS — correspondance des objectifs de recherche aux services de séquençage
Matrice de sélection d'application pour quatre principales approches NGS—WGS, WES, RNA-seq et métagénomique—montrant les cas d'utilisation optimaux, les variables de conception, les risques et la profondeur de lecture recommandée pour chaque type d'application.
Choisir entre Amplicon, Panel, Exome, Génome et Transcriptome
Le choix entre ces cinq types de tests dépend de l'étendue de la question de recherche et du potentiel de découverte requis.
| Option | Meilleur pour | Potentiel de découverte | Complexité des données | Principale limitation |
|---|---|---|---|---|
| Amplicon | Régions petites connues, grands nombres d'échantillons | Bas | Faible–Moyen | Portée étroite |
| Panneau cible | Ensembles de gènes connus | Moyen | Moyen | Dépendant du design |
| WES | Variantes de codage | Moyen–Élevé | Moyen–Élevé | Manque des régions non codantes |
| WGS | Découverte à l'échelle du génome | Élevé | Élevé | Charge de données plus élevée |
| RNA-seq | Expression / transcriptome | Élevé pour le niveau d'ARN | Élevé | sensible à la qualité de l'ARN |
Logique de décision d'achat : Le cible est-elle connue ou inconnue ? La découverte est-elle nécessaire ou la confirmation est-elle suffisante ? Une couverture génomique est-elle nécessaire ou une région ciblée est-elle adéquate ? La question porte-t-elle sur l'ADN ou l'ARN ? La qualité de l'échantillon et le budget soutiennent-ils la complexité de l'approche choisie ? Répondre à ces questions avant de sélectionner un type de test permet d'éviter des changements coûteux en cours de projet.
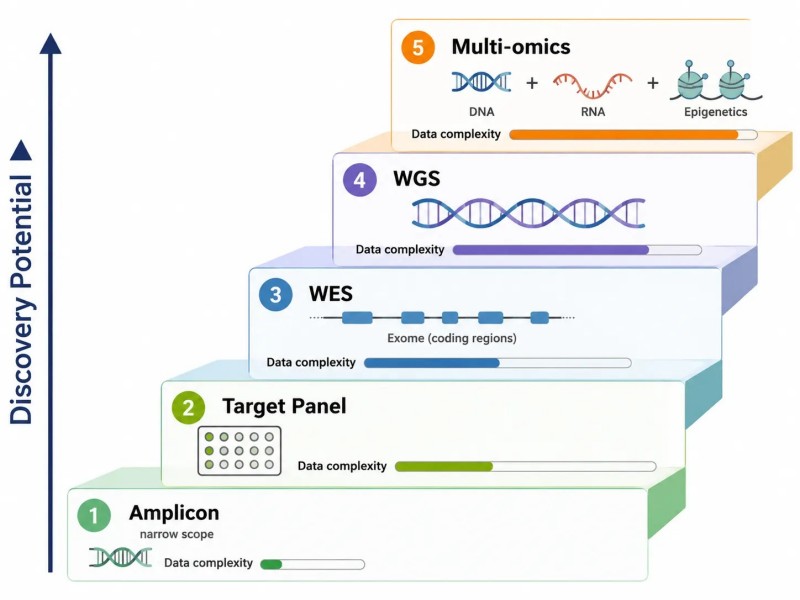 Figure 10. Échelle de sélection des tests NGS — de l'amplicon au génome entier
Figure 10. Échelle de sélection des tests NGS — de l'amplicon au génome entier
Ladder de sélection d'essai NGS à cinq niveaux, allant de l'amplicon (potentiel de découverte le plus faible, portée la plus étroite) à la WGS (potentiel de découverte le plus élevé, couverture la plus large), avec une logique de décision pour choisir l'essai approprié en fonction des connaissances sur la cible et des exigences de recherche.
Causes courantes d'échec des projets NGS — et comment les prévenir
Comprendre les modes de défaillance avant de commencer un projet est la stratégie de prévention la plus efficace. Les échecs peuvent survenir à chaque étape.
| Problème observé | Cause possible | Prévention au niveau de la conception |
|---|---|---|
| Faibles lectures utilisables | Contamination, dimères d'adaptateurs, faible qualité de la bibliothèque | Contrôle qualité et nettoyage avant séquençage |
| Couverture inégale | biais GC, biais de capture, biais de fragmentation | Ajustement de la stratégie de plateforme et de bibliothèque |
| Haute duplication | ADN à faible entrée, sur-PCR | Surveillance de la complexité des bibliothèques, protocoles sans PCR |
| Signal biologique faible | Mauvaise conception de réplicats, effets de lot | Conception statistique avant le séquençage |
| Mauvaise annotation | Inadéquation de la base de données, référence obsolète | Sélection de base de données et contrôle de version |
Échecs au niveau de l'échantillon : Dégradation, contamination, entrée insuffisante ou méthode d'extraction médiocre — à traiter par un contrôle qualité rigoureux avant le séquençage.
Échecs au niveau de la bibliothèque : Contamination par dimères d'adaptateurs, faible efficacité de conversion, sur-amplification, fragmentation biaisée — à traiter en optimisant les protocoles de bibliothèque et en incluant des points de contrôle de qualité.
Échecs au niveau du séquençage : Sous-chargement ou surcharge de la cellule de flux, bibliothèques à faible diversité entraînant des échecs de calibration, déséquilibre entre les échantillons multiplexés, profondeur insuffisante — à traiter par des calculs de chargement précis et une planification de la diversité.
Échecs au niveau des données : Taux de cartographie faible, choix de génome de référence médiocre, forte duplication, effets de lot, incompatibilité de base de données — à traiter en incluant des contrôles et en planifiant l'analyse avant le séquençage.
Échecs de conception de projet : La question de recherche est trop large pour l'approche choisie, plateforme ou type d'essai inapproprié, pas de réplicats biologiques, attentes en aval irréalistes — à traiter en utilisant le cadre de ce guide avant de consacrer des ressources.
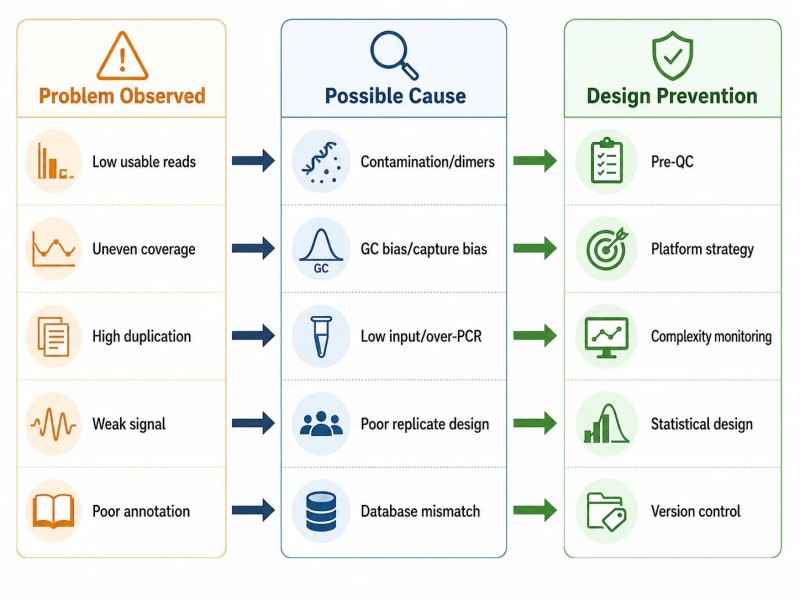 Figure 11. Carte de dépannage NGS — problème, cause et prévention
Figure 11. Carte de dépannage NGS — problème, cause et prévention
Légende : Carte complète de dépannage NGS couvrant cinq niveaux de défaillance : échantillon, bibliothèque, séquençage, données et conception de projet, avec les problèmes observés, les causes possibles et les stratégies de prévention au niveau de la conception pour chaque catégorie.
Comment préparer une demande de projet pour un fournisseur de services NGS
Une demande de projet bien préparée accélère le processus de consultation et réduit le risque d'attentes mal alignées. Les informations suivantes doivent être préparées avant de contacter un fournisseur de services de séquençage.
Informations de base à fournir : Espèce et type d'échantillon, ADN ou ARN, numéro d'échantillon, méthode d'extraction, concentration et quantité totale, données d'intégrité (RIN, DV200 ou image de gel), objectif de recherche et livrables attendus. Inclure ces informations dans la demande initiale permet au prestataire de services d'évaluer la faisabilité et de recommander une stratégie appropriée sans avoir besoin de clarifications répétées.
Informations sur la conception du projet : Que ce soit un génome de référence disponible, que l'étude cible des régions connues ou nécessite une découverte, que l'appel de variants, le profilage d'expression, l'assemblage ou l'annotation soient nécessaires, que des réplicats biologiques soient inclus, et les informations sur les lots.
Questions à poser au prestataire de services : Quelle stratégie NGS est recommandée et pourquoi ? Quels indicateurs de qualité seront rapportés à chaque étape ? Quels sont les livrables bruts, propres et finaux ? Comment les résultats de contrôle qualité anormaux sont-ils gérés ? Le pipeline bioinformatique peut-il être personnalisé pour le projet ?
Formuler des questions de manière constructive : Au lieu de "Pouvez-vous garantir le succès ?", demandez "Quels facteurs d'échantillonnage ou de conception affectent la qualité des données dans ce type de projet ?" Au lieu de "À quelle vitesse pouvez-vous livrer ?", demandez "Quels sont les principaux points de contrôle de la qualité dans le calendrier du projet ?"
 Figure 12. Liste de contrôle pour l'enquête sur le projet NGS - informations à préparer et questions à poser
Figure 12. Liste de contrôle pour l'enquête sur le projet NGS - informations à préparer et questions à poser
Liste de contrôle pour la préparation de la demande de projet montrant des informations de base (espèce, type d'échantillon, méthode d'extraction, concentration, données d'intégrité), des informations sur la conception du projet (génome de référence, découverte vs ciblé, réplicats) et des questions constructives à poser à un fournisseur de services de séquençage.
Liste de vérification pour la sélection de la stratégie NGS — Un cadre en 10 étapes
- Définir la question biologique.
- Identifier si la cible est l'ADN, l'ARN, épigénétique, microbien ou unicellulaire.
- Confirmer la qualité de l'échantillon et la faisabilité de l'entrée.
- Sélectionnez le type de test : amplicon / panel / WES / WGS / RNA-seq / métagénomique
- Sélectionnez la plateforme de séquençage : lecture courte / lecture longue / hybride
- Définir la longueur de lecture, la profondeur et les attentes en matière de couverture.
- Confirmer la stratégie de préparation de bibliothèque
- Définir des indicateurs de qualité (QC) à suivre avant le séquençage.
- Définir les livrables en bioinformatique
- Préparez les informations du projet pour la consultation.
Suivre cette liste de contrôle de manière systématique minimise le risque de corrections coûteuses en cours de projet et garantit que la stratégie de séquençage est alignée avec les objectifs de recherche dès le départ.
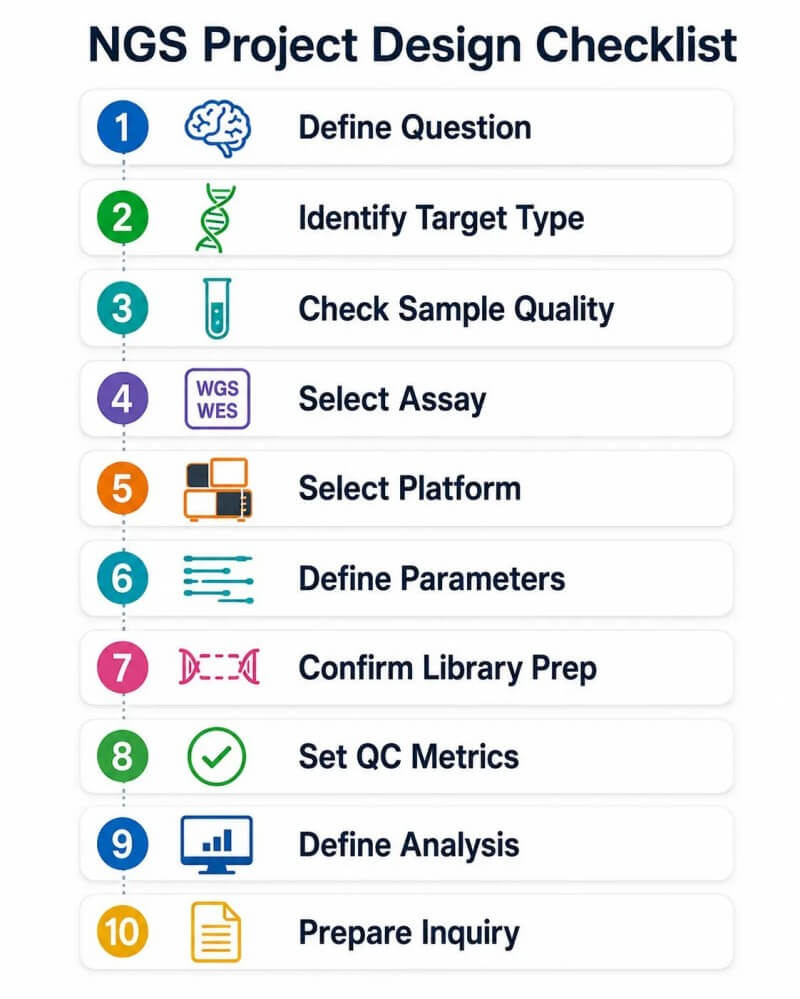 Figure 13. Liste de contrôle de la stratégie NGS pour les projets de recherche — Cadre de conception en 10 étapes
Figure 13. Liste de contrôle de la stratégie NGS pour les projets de recherche — Cadre de conception en 10 étapes
Légende : Cadre de sélection de stratégie NGS en 10 étapes, allant de la définition de la question biologique à la sélection du type d'essai, de la plateforme de séquençage, de la longueur de lecture, de la profondeur, de la stratégie de bibliothèque, des métriques de QC, des livrables en bioinformatique et de la préparation d'une demande de projet pour consultation.
Conclusion — La valeur du NGS réside dans l'adéquation entre la stratégie et la question.
Le séquençage de nouvelle génération est un système de projet à variables multiples. La plateforme, la bibliothèque, la profondeur, la qualité de l'échantillon et le pipeline d'analyse contribuent tous à la capacité d'interprétation des données finales. Pour les chercheurs évaluant les options de NGS, les questions les plus importantes ne portent pas sur ce qu'est le NGS, mais sur la stratégie qui correspond le mieux à l'objectif de recherche, si l'échantillon soutient l'approche choisie, quelles métriques de données doivent être définies à l'avance, et si les livrables en bioinformatique peuvent répondre à la question biologique initiale.
Pour la planification de projets à des fins de recherche, les chercheurs peuvent préparer le type d'échantillon, l'objectif de recherche, les résultats d'analyse attendus et les informations de contrôle qualité disponibles avant de discuter d'une stratégie NGS avec leur prestataire de services choisi.
Les projets NGS les plus réussis sont ceux où la conception expérimentale est guidée par la question biologique, la qualité de l'échantillon est évaluée avant le début du séquençage, la plateforme et la profondeur sont choisies en fonction du type de variant cible et des caractéristiques du génome, et l'analyse bioinformatique est planifiée comme une partie intégrante du projet plutôt que comme une réflexion après coup. En appliquant le cadre décrit dans ce guide, les chercheurs peuvent réduire considérablement le risque de corrections coûteuses en cours de projet et s'assurer que leur investissement en séquençage produit des résultats interprétables et prêts pour publication.
FAQ
Quelle est la différence entre la profondeur de séquençage et la couverture ?
La profondeur fait référence au nombre moyen de lectures couvrant chaque base dans la région cible. La couverture peut désigner soit la fraction du génome couverte par au moins une lecture (largeur), soit l'uniformité de la profondeur à travers le génome. Les deux métriques sont nécessaires pour évaluer la qualité des données.
Puis-je combiner le séquençage à court et à long reads dans un même projet ?
Oui. Les stratégies hybrides qui utilisent des lectures longues pour la continuité et des lectures courtes pour le polissage sont standard pour l'assemblage de novo et la détection de variantes structurelles. De nombreux assemblages de génomes publiés utilisent cette approche combinée.
Quelle est la quantité minimale d'ADN requise pour le séquençage de nouvelle génération (NGS) ?
L'entrée minimale varie selon la méthode de préparation de la bibliothèque : les kits standard basés sur la PCR fonctionnent avec 0,1 ng à 1 µg, les kits sans PCR nécessitent 100 ng à 1 µg, les kits basés sur la tagmentation fonctionnent avec 1-50 ng, et les kits à ultra faible entrée peuvent fonctionner avec aussi peu que 50 pg. Choisir le kit approprié en fonction de l'entrée disponible est crucial.
Comment évaluer la qualité des données NGS à partir d'un rapport de séquençage ?
Métriques clés à vérifier : pourcentage Q30 (>85% pour de bonnes analyses), taux de cartographie (>80% pour l'ADN humain), taux de duplication (<15% pour le WGS), contenu en adaptateurs (<1% après découpe), et taux cible pour les méthodes basées sur la capture. Un bon rapport de QC devrait inclure toutes ces métriques avec des explications claires.
Comment choisir entre le séquençage du génome entier (WGS) et le séquençage de l'exome (WES) pour un projet de génétique humaine ?
Choisissez le séquençage génomique complet (WGS) lorsque la détection complète des variants (y compris les variants non codants, structurels et régulateurs) est nécessaire et que le budget le permet. Choisissez le séquençage des exomes (WES) lorsque l'accent est mis sur les variants codants et que le projet nécessite une profondeur plus élevée dans les régions exoniques à un coût global inférieur. Le WES manque environ 98 % du génome, y compris la plupart des régions régulatrices et introniques, qui sont de plus en plus reconnues comme importantes dans la génétique des maladies complexes.
Pour usage de recherche uniquement.
Références:
- Aperçu du flux de travail NGS d'Illumina. Illumina, Inc.
- Évaluation des performances des plateformes de séquençage ADN dans l'étude de séquençage de nouvelle génération de l'ABRF. Biotechnologie de la nature. 2021;39:1348-1365.
- La chimie du séquençage de nouvelle génération. Biotechnologie de la nature2023;41:1709-1715.
- Technologie de séquençage par nanopores, bioinformatique et applications. Biotechnologie de la Nature. 2021;39:1348-1365.
- Séquençage de longues lectures par consensus circulaire précis. Biotechnologie de la nature2019 ; 37 : 1155-1162.


