
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Pipeline d'analyse des données de séquençage bisulfite de génome entier (WGBS)
Introduction
La méthylation de l'ADN est un composant important de l'épigénétique et joue un rôle essentiel dans le maintien de la fonction cellulaire normale, l'empreinte génétique, le développement embryonnaire et la tumorigénèse humaine. Séquençage bisulfite de l'ensemble du génome (WGBS) convertit la cytosine non méthylée (C) en thymine (T) grâce au traitement au bisulfite sur l'ADN génomique. Par la suite, un resequencement du génome entier de l'ADN traité et une comparaison avec le génome de référence ont été réalisés pour atteindre une résolution à base unique et une analyse de niveau de méthylation de haute précision au niveau génomique. Le WGBS est largement utilisé dans les études de différenciation cellulaire, de développement des tissus, ainsi que dans l'élevage animal et végétal, la santé humaine et les maladies.
Pipeline d'analyse des données de WGBS
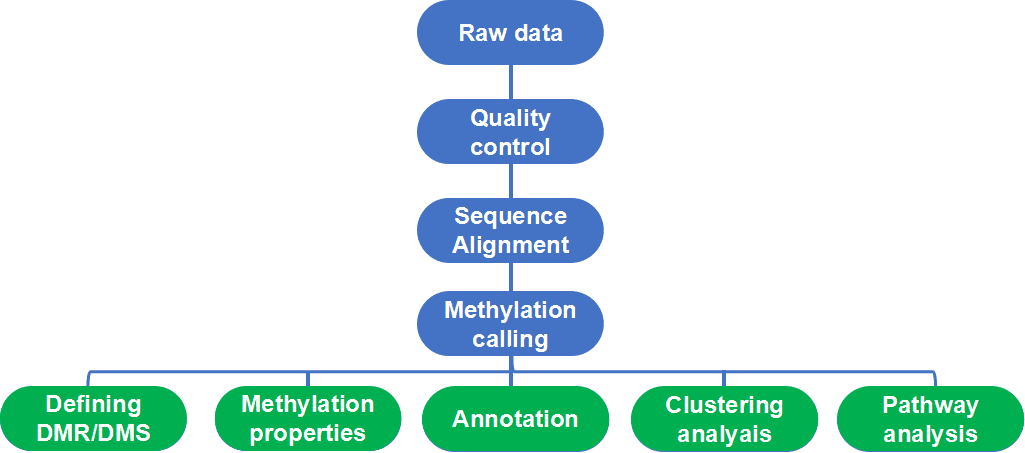 Figure 1. Flux de travail et analyse bioinformatique de base du WGBS.
Figure 1. Flux de travail et analyse bioinformatique de base du WGBS.
- Contrôle de qualité
Le contrôle de qualité des lectures brutes est une étape essentielle pour garantir que les données sont de haute qualité et adaptées à une analyse ultérieure. Les données brutes peuvent être contaminées par des amorces PCR et des dimères lors de la construction de la bibliothèque ou altérées par des bases de faible qualité lors du séquençage. Par conséquent, le retrait des adaptateurs et le découpage/filtrage des lectures sont nécessaires pour éliminer les bases de faible qualité de l'analyse suivante.
De nombreux programmes, tels que FastQC, BIGpre et PIQA, peuvent fournir diverses analyses pour le contrôle de la qualité. Parmi eux, FastQC est l'outil le plus reconnu pour le contrôle de la qualité. Il traite de nombreux aspects des lectures brutes et génère un rapport HTML qui résume chaque contrôle de qualité effectué. En général, les résultats couvrent différents aspects de la qualité des lectures brutes, tels que le nombre total de lectures, la distribution de la qualité des lectures, la distribution du contenu en GC, la qualité générale des séquences et bien plus encore.
- Cartographie des lectures sur le génome de référence
Ensuite, les lectures doivent être alignées sur le génome de référence. Les outils d'alignement conventionnels tels que BWA, Bowtie ou Maq ne sont pas adaptés en raison de la dissimilarité entre les génomes de référence standard et les lectures de l'ADN traité au bisulfite. Il existe de nombreux outils disponibles, tels que Bismark, BS-Seeker2, Bison, Last ou B-SOLANA. Les outils d'alignement représentés par Bismark utilisent des aligners de courtes lectures existants et gèrent les exigences uniques à l'analyse des données WGBS en interne, en convertissant C en T dans les lectures séquencées et le génome de référence avant le mapping. D'autres méthodes, comme Last, utilisent une matrice de score spécifique qui peut tolérer les discordances C-T.
- Appel de méthylation
Les doublons PCR peuvent être identifiés de manière computationnelle après alignement au génome de référence, car ils gonflent erronément les couvertures génomiques et entraîneront des erreurs de faux positifs dans les analyses ultérieures. En général, la manière dont nous résolvons le biais PCR est de supprimer les lectures qui sont alignées à la même position sur le même brin du génome de référence. L'appel de méthylation peut être effectué à l'aide d'outils d'alignement tels que Bismark, qui fournit des fichiers BAM, ainsi que des métriques et des fichiers supplémentaires liés à l'appel de méthylation.
Une transformation incomplète et une dégradation de l'ADN peuvent se produire lors du traitement au bisulfite, c'est-à-dire que les Cs non méthylés ne sont pas entièrement convertis en Ts. Par conséquent, un contrôle de qualité doit encore être effectué après l'alignement et l'appel de méthylation pour éliminer les erreurs de faux positifs. Il existe deux façons de calibrer le taux de conversion ; l'une consiste à utiliser le pourcentage de méthylation non-CpG, l'autre consiste à utiliser des Cs non méthylés pour ajouter des séquences de spike-in et calculer le nombre de Ts.
Tableau 1. Logiciel d'appel DMR (Condon) et al.. 2018).
| Programme | Identification DMR | Exécution |
| Défiant | Expansion de Welch pondérée | Binaire |
| BSmooth | Lissage par vraisemblance locale avec test binomial | R |
| methylKit | Test exact de Fisher ou régression logistique avec carrelage | R |
| MethylSig | bêta-binomial | R |
| Méthylène | pvaleur par binomiale bêta | Binaire |
| MOABS | bêta-binomial | Binaire |
| RADMeth | régression bêta-binomiale | Binaire |
L'analyse bioinformatique et la visualisation du WGBS comprennent des analyses de données de base telles que l'analyse du niveau et de la densité de méthylation, l'analyse des motifs et des analyses de données avancées telles que l'analyse des réseaux de co-expression.
- Identification des régions différemment méthylées
L'analyse de méthylation différentielle vise à identifier des régions de méthylation différentielle (DMR) entre des groupes spécifiques. Les DMS (sites CpG méthylés de manière différentielle) font référence à des sites de cytosine présentant des niveaux de modification de méthylation significativement différents entre différents échantillons. Cela reflète la différence dans la modification de méthylation entre les échantillons à une résolution de base unique et constitue une unité de base pour étudier l'expression des gènes régulés par la méthylation.
- Segmentation du méthylome
L'analyse des dynamiques de méthylation est également importante au sein de l'échantillon. Par exemple, des dépressions dans les profils de méthylation indiquent généralement des régions régulatrices, et de nombreuses régions du corps des gènes sont largement méthylées et pauvres en CpG. Ces observations peuvent décrire les structures et la fonction des gènes. Les états de méthylation (y compris les régions faiblement méthylées, les régions entièrement méthylées et les régions non méthylées) peuvent être identifiés par la segmentation du méthylome à l'aide d'approches informatiques, telles que MethylSeekR et MethPipe.
- Annotation de MDR/DMS et segments
Les DMR/DMS et les segments doivent souvent être interprétés en termes biologiques avec une base de données d'annotation du génome. Il existe plusieurs outils capables de produire de telles annotations. Par exemple, le package Bioconductor genomation peut être utilisé pour annoter les DMR/DMS et intégrer les proportions de méthylation. CHIPpeakAnno, un autre package similaire, est conçu non seulement pour l'annotation des pics ChIP-seq, mais aussi pour l'annotation des DMR/DMC dans une certaine mesure.
- Analyse de regroupement
Le regroupement fait référence à la classification d'objets en grappes en fonction de leur similarité. L'analyse de regroupement est importante pour identifier les sous-types moléculaires de maladies. L'analyse PCA, également connue sous le nom d'analyse en composantes principales, est une méthode courante pour détecter des motifs dans les données de méthylation entre différents traitements ou différentes étapes de développement, expliquant ainsi la variation des motifs de méthylation dans différents traitements. L'analyse PCA détecte également les effets de lot. Une autre méthode courante pour l'analyse de regroupement est le regroupement hiérarchique. Des outils tels que HumMeth27QCReport et methylkit offrent à la fois des options d'analyse PCA et de regroupement hiérarchique en utilisant des données normalisées. M-valeurs.
- Analyse des voies
Selon les données de méthylation de chaque échantillon, une analyse de regroupement et une analyse d'enrichissement ont été réalisées pour analyser la similarité entre les échantillons. Cela inclut généralement une analyse d'enrichissement GO et une analyse des voies KEGG, toutes deux utilisant le serveur web DAVID.
Analyse GOL'Ontologie des gènes (GO) est un système de classification standard international pour la fonction des gènes. Réaliser une analyse d'enrichissement GO sur des gènes connexes peut explorer les fonctions biologiques, la composition cellulaire et les processus biologiques des gènes régulés par les DMR.
Analyse de voie KEGG. L'analyse d'enrichissement significatif des voies KEGG peut déterminer les voies métaboliques biochimiques et les voies de transduction du signal les plus importantes impliquées dans les gènes régulés par les DMR.
Chez CD Genomics, nous sommes dédiés à fournir des services fiables. séquençage épigénomique services, y compris séquençage bisulfite ciblé, séquençage bisulfite à représentation réduite (RRBS), séquençage bisulfite de tout le génome, Séquençage MeDIP, et ChIP-seqSi vous êtes intéressé par nos services, n'hésitez pas à nous contacter.
Références :
- De Carvalho D.D. et al. Le dépistage de la méthylation de l'ADN identifie des événements épigénétiques moteurs de la survie des cellules cancéreuses. Cellule cancéreuse, 2012, 21(5):655-667
- Jeong M., Goodell M. A. Nouvelles réponses à de vieilles questions grâce aux cartes génomiques de la méthylation de l'ADN dans les cellules hématopoïétiques. Exp Hematol, 2014, 42(8) : 609-617
- Kulis M., Esteller M. Méthylation de l'ADN et cancer. Adv Genet, 2010 70:27-56
- Wu H., Xu T., Feng H. et al.Détection des régions différemment méthylées à partir de données de séquençage bisulfite du génome entier sans réplicats. Acides Nucleiques Res, 2015, 43(21):132-141.
- Swarnaseetha A., Aspects méthodologiques de l'analyse du séquençage bisulfite de génome entier. Briefings in Bioinformatics, 2015, (1) : 369-379.
- Condon D.E., et al.Défiant : (DMRs : identification et annotation faciles et rapides) identifie les régions méthylées différemment du hippocampe de rat en déficit en fer. BMC bioinformatique, 2018, 19(1) : 31.
- Wreczycka K, et al.Stratégies pour analyser les données de séquençage au bisulfite. Journal de biotechnologie, 2017, 261 : 105-115.


