
 Directives de Soumission d'Échantillons
Directives de Soumission d'Échantillons
Analyse de la Variation du Nombre de Copies (CNV) : Méthodes de Détection, Stratégies de Profondeur et Outils de Bioinformatique
La variation du nombre de copies (CNV) fait référence à la duplication ou à la suppression de segments d'ADN de plus de 1 kb. Dans le génome humain, les CNV représentent plus de différences totales en paires de bases entre les individus que les variants de nucléotides uniques, mais ils restent plus difficiles à détecter car le signal principal—la profondeur de lecture—est une variable continue affectée par de nombreux facteurs techniques confondants. Contrairement à l'appel de SNV, qui dépend de l'identité de la base à une position unique, la détection de CNV nécessite d'intégrer les informations de profondeur à travers des fenêtres génomiques, de corriger les biais systématiques et de segmenter le génome en régions de nombre de copies cohérent.
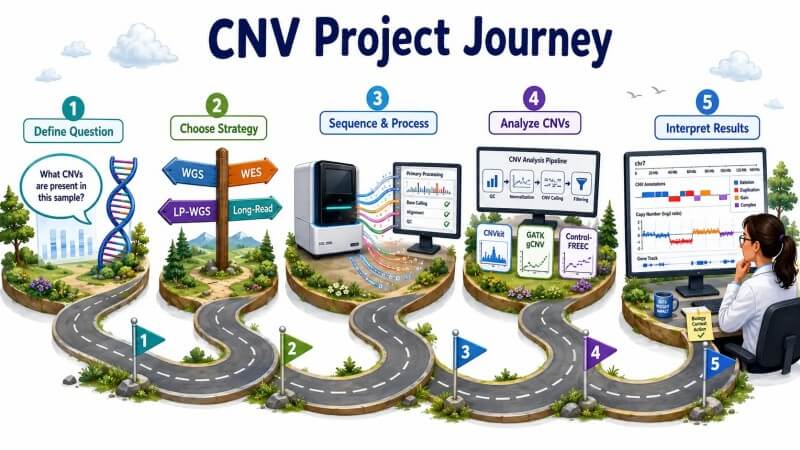
Ce guide fournit un cadre pratique pour les chercheurs ayant de l'expérience avec les données NGS et qui doivent concevoir, exécuter et interpréter des projets d'analyse de CNV. Il couvre la base algorithmique de la détection de CNV par profondeur de lecture, la relation quantitative entre la profondeur de séquençage et la sensibilité de détection, une comparaison détaillée des outils bioinformatiques avec des données de performance de référence, les principaux défis techniques qui compromettent l'exactitude, et des stratégies pratiques pour la conception de projets - de dépistages à faible profondeur à un profilage génomique haute résolution. L'accent est mis tout au long sur la manière dont les décisions analytiques affectent les types et les tailles de CNV qui peuvent être détectés de manière fiable, et sur l'évitement des pièges courants qui conduisent à des résultats faussement positifs ou faussement négatifs. Que vous analysiez des CNV germinaux dans une étude de population de plus de 1 000 échantillons ou des CNV somatiques dans une petite cohorte de cancer, les principes de normalisation GC, de filtrage de mappabilité et de segmentation consciente de la cohorte s'appliquent à toutes les échelles d'analyse de CNV.
Services de séquençage du génome entier soutenir la détection des CNV dans toutes les configurations de profondeur—de l'analyse germinale à haute couverture de 30× à le dépistage somatique à faible couverture de 0,5-2×—avec des pipelines bioinformatiques adaptés à chaque approche.
Qu'est-ce que la variation du nombre de copies et pourquoi est-ce important ?
Les variants du nombre de copies sont des altérations structurelles dans lesquelles un segment d'ADN est présent en plus ou moins de copies que le génome de référence. Ils varient d'environ 1 kb à plusieurs mégabases, et leur formation est principalement entraînée par la recombinaison homologue non allèlique (NAHR) entre des duplications segmentaires flanquantes pendant la méiose, et par la jonction des extrémités non homologues (NHEJ) dans des contextes méiotique et mitotique. Les CNV sont classés en délétions (perte d'un segment génomique, réduisant le nombre de copies à 1 ou 0) et en duplications (gain d'une copie supplémentaire, augmentant le nombre de copies à 3 ou plus). Cette classification est détectable à partir des données de séquençage par l'ampleur et la direction du changement de profondeur de lecture : une délétions hétérozygote réduit la profondeur attendue de 50 %, tandis qu'une délétions homozygote la réduit à zéro.
L'impact biologique des CNVs est substantiel. Dans la lignée germinale, les CNVs sont une cause bien établie de troubles génétiques : le syndrome de microdélétion 22q11.2 (1 sur 4 000 naissances vivantes), la maladie de Charcot-Marie-Tooth (duplication de PMP22) et le syndrome de Smith-Magenis (délétion de RAI1) en sont des exemples classiques. Des études à l'échelle de la population estiment que de grands CNVs (>50 kb) affectent environ 15 % du génome en nombre de copies et représentent plus de variation génétique interindividuelle que tous les SNVs combinés. Dans le cancer, les amplifications focales d'oncogènes tels que MYC, EGFR, KRAS et ERBB2 entraînent directement la progression tumorale, tandis que les délétions homozygotes ou hétérozygotes de gènes suppresseurs de tumeurs, y compris TP53, CDKN2A, PTEN et RB1, éliminent des voies régulatrices critiques. Services d'appel de variants inclure la détection des CNV comme un composant standard de l'analyse génomique complète.
Comment la détection de CNV à partir des données de séquençage fonctionne — La base algorithmique
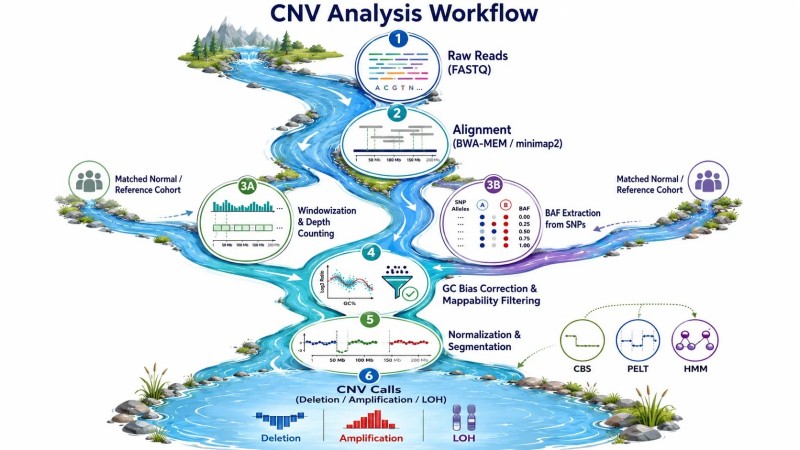
Toutes les méthodes de détection des CNV basées sur le séquençage partagent un cadre algorithmique commun reposant sur l'analyse de la profondeur de lecture, bien que les mises en œuvre spécifiques diffèrent considérablement entre les outils. Comprendre ce cadre est essentiel pour interpréter les résultats des CNV et résoudre les analyses échouées.
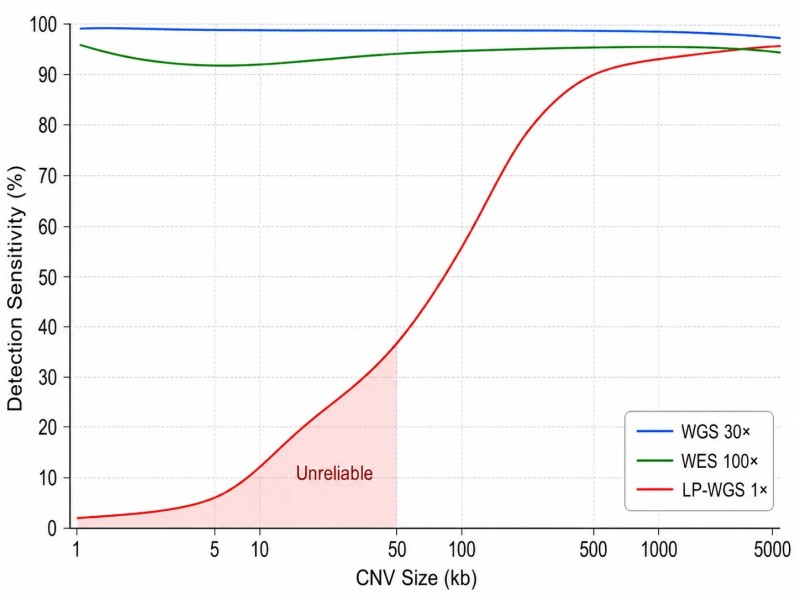
Lecture de la profondeur, comptage et fenêtrageLes lectures séquencées sont alignées sur le génome de référence, et le nombre de lectures s'alignant sur chaque fenêtre génomique est compté. La taille de la fenêtre est un paramètre critique : des fenêtres plus petites (100 pb à 1 kb) offrent une résolution de rupture plus élevée mais une puissance statistique par fenêtre plus faible, tandis que des fenêtres plus grandes (10-100 kb) augmentent le rapport signal sur bruit au prix de ruptures floues. Pour le WGS à 30×, des fenêtres de 1 kb fournissent une puissance suffisante pour la détection de CNV. Pour le LP-WGS à 1×, des fenêtres de 100 à 500 kb sont nécessaires. La profondeur de lecture dans chaque fenêtre suit une distribution de Poisson avec une moyenne égale à la couverture attendue, et les CNV sont identifiés comme des régions où la profondeur observée s'écarte significativement de cette attente après normalisation.
Normalisation — suppression de la variation techniqueLa profondeur de lecture brute est dominée par des facteurs techniques sans rapport avec le nombre de copies. Le contenu en GC produit à lui seul une plage de profondeur de couverture de 2 à 5 fois—les régions riches en GC se séquencent avec une efficacité plus élevée dans la plupart des protocoles de préparation de bibliothèques, créant des pics et des vallées systématiques qui sont cohérents entre les échantillons d'une même course de séquençage. La normalisation corrige cela en calculant le rapport entre la profondeur observée et la profondeur attendue pour des fenêtres de contenu en GC similaire, puis en lissant la courbe de biais en GC pour éliminer le bruit à petite échelle. La correction de la mappabilité exclut les fenêtres où une fraction substantielle du génome ne peut pas être mappée de manière unique par de courtes lectures—typiquement les centromères, les télomères et les duplications segmentaires. Sans ces corrections, le signal de biais en GC produirait de fausses détections de CNV dans la plupart des régions riches et pauvres en GC du génome.
SegmentationAprès normalisation, le profil de profondeur est divisé en segments de nombre de copies cohérent à l'aide d'algorithmes de segmentation. La segmentation binaire circulaire (CBS), mise en œuvre dans le package R DNAcopy, divise récursivement le génome en segments en testant des fenêtres adjacentes pour des différences significatives dans la profondeur moyenne. L'algorithme PELT (Pruned Exact Linear Time), utilisé par GATK gCNV, est plus rapide et évolue de manière linéaire avec le nombre de fenêtres. Les modèles de Markov cachés (HMM), utilisés par XHMM pour les données d'exome, traitent l'état du nombre de copies comme une variable cachée déduite de la séquence de profondeur observée. Le choix de l'algorithme de segmentation influence l'équilibre entre la sensibilité (détection de petits CNV) et la spécificité (éviter de trop diviser le génome en de nombreux petits segments qui reflètent le bruit plutôt que de véritables CNV). En pratique, CBS produit des appels plus conservateurs avec moins de faux positifs, tandis que les HMM sont plus sensibles au prix d'une augmentation des taux de faux positifs pour les événements à fenêtre unique.
Attribution de génotype à partir de la fréquence de l'allèle BEn plus de la profondeur de lecture, les positions de SNP hétérozygotes dans les lectures alignées fournissent des informations sur la fréquence de l'allèle B (BAF). Dans les régions diploïdes, le BAF se regroupe autour de 0,5 pour les SNP hétérozygotes. Dans les régions avec changement du nombre de copies, le BAF s'écarte de 0,5 : la perte d'hétérozygotie (LOH) déplace le BAF vers 0 ou 1, tandis que l'amplification le déplace selon un modèle qui dépend du ratio des copies d'allèles. Des outils tels que Control-FREEC et Canvas intègrent le BAF avec la profondeur pour distinguer les types de CNV (LOH neutre en copie vs. véritable délétion) et pour détecter les CNV dans des échantillons avec contamination cellulaire normale, où le signal de profondeur seul peut être ambigu.
La relation quantitative entre la profondeur et la force du signal CNV.La confiance d'un appel de CNV dépend du rapport signal sur bruit de la déviation de profondeur observée. Pour une délétion hétérozygote, la réduction de profondeur attendue est de 50 %. L'écart-type de la profondeur de lecture dans une fenêtre est d'environ sqrt(profondeur_moyenne) pour des comptages distribués selon une loi de Poisson. À 30× WGS avec des fenêtres de 1 kb contenant environ 30 lectures, l'écart-type attendu est d'environ 5,5 lectures (18 %), ce qui rend une réduction de 50 % environ 9 écarts-types de la moyenne — facilement détectable. À 1× LP-WGS avec des fenêtres de 200 kb contenant ~200 lectures, l'écart-type attendu est d'environ 14 lectures (7 %), et le même CNV produit une réduction de 50 % détectable à ~7 écarts-types. Cependant, après que la correction GC et la normalisation aient éliminé le biais systématique, le bruit résiduel dans le LP-WGS est environ 2-3 fois plus élevé que l'attente de Poisson en raison de la variabilité de fragmentation et des artefacts d'alignement. Ce bruit supplémentaire est la raison pour laquelle le LP-WGS nécessite des fenêtres plus grandes que le WGS pour une sensibilité de détection équivalente. Comprendre cette relation quantitative aide les chercheurs à établir des attentes réalistes pour la détection des CNV — il existe un compromis direct entre la taille du CNV, la profondeur de séquençage et la confiance de détection qui ne peut pas être surmonté par une amélioration de la normalisation bioinformatique seule.
Figure 1 : Quatre approches de détection des CNV — couverture génomique, résolution et profondeur optimale

Détection des CNV basée sur le séquençage — Comparaison de quatre approches
La détection des CNV à partir des données de séquençage peut être réalisée en utilisant quatre approches qui diffèrent fondamentalement en termes de couverture génomique, de profondeur et de coût. Le choix entre elles détermine quels types et tailles de CNV peuvent être détectés.
WGS à une couverture de 30×Le séquençage de l'ADN à l'échelle du génome à une profondeur standard fournit la détection de CNV la plus complète. La profondeur de lecture est mesurée sur l'ensemble du génome dans des fenêtres de 100 pb à 1 kb, corrigée pour le contenu en GC et la cartographie, et segmentée pour identifier les régions avec une couverture significativement décalée. Le séquençage du génome entier à 30× détecte des délétions hétérozygotes aussi petites que 1-5 kb et des amplifications aussi petites que 5-10 kb, couvrant à la fois les régions codantes et non codantes. Le compromis est le coût du séquençage—environ 90-100 Gb par génome—ce qui limite le débit d'échantillons pour les études de grandes cohortes. Au sein de la fraction codante du génome, la résolution est généralement de 1-2 kb, suffisante pour détecter des CNV à exon unique.
WES à 100-200×Le séquençage de l'exome entier ne capture que la fraction codante du génome (~1-2%, environ 35 Mb), mais la profondeur de lecture plus élevée offre une meilleure puissance statistique pour la détection des CNV dans les régions capturées à un coût comparable à celui du WGS. Le principal défi de la détection des CNV basée sur le WES est la couverture non uniforme inhérente à la capture ciblée : l'efficacité d'hybridation varie entre les sondes et entre les régions au sein du même ensemble de sondes, introduisant un bruit systématique qui est spécifique à l'échantillon et ne peut pas être entièrement corrigé par une normalisation GC générique. ECOLE (2023, Nature Communications), un appelant de CNV basé sur l'apprentissage profond pour les données WES, aborde ce problème en entraînant un réseau de neurones convolutifs sur des données simulées qui intègrent le profil de bruit spécifique à la capture de chaque kit, atteignant 20-30% de faux positifs en moins que les appelants de CNV WES conventionnels. Pour les chercheurs utilisant le WES pour l'analyse des CNV, une couverture cible moyenne d'au moins 100× est recommandée, avec au moins 30 échantillons normaux inclus dans le projet pour la construction de références. Services de séquençage de l'exome entier offrir une couverture de 150-200× pour un design d'étude WES optimisé pour la CNV.
LP-WGS à 0,5-5×Le séquençage du génome entier à faible profondeur (LP-WGS) séquence l'ensemble du génome à une fraction de la profondeur standard, ce qui en fait la méthode de dépistage CNV la plus rentable. Avec une couverture de 1×, environ 3 Gb par échantillon, le LP-WGS détecte les CNV supérieurs à 50-100 kb avec une sensibilité comparable à celle des microarrays chromosomiques, ce qui en fait une alternative viable pour le dépistage clinique des CNV lorsque les exigences de résolution sont modérées. Un repère de 2025 dans les Briefings in Bioinformatics a démontré qu'à 1× avec des fenêtres de 200 kb, le LP-WGS atteint une sensibilité >90 % pour les délétions >100 kb et >85 % pour les duplications >150 kb. Le paramètre de taille de fenêtre est le levier clé : des fenêtres plus grandes améliorent la sensibilité au détriment de la résolution des points de rupture, et le réglage optimal varie inversement avec la profondeur (200 kb à 1×, 50 kb à 5×). Pour les projets qui doivent équilibrer la détection des CNV avec le débit d'échantillons, le LP-WGS offre la meilleure rentabilité par échantillon.
Séquençage long (PacBio HiFi / Nanopore)Des lectures longues s'étendant sur 10-20 kb (HiFi) ou dépassant 100 kb (Nanopore) abordent la détection des CNV selon un principe fondamentalement différent : au lieu d'inférer le nombre de copies à partir de la profondeur, elles peuvent physiquement couvrir les points de rupture des CNV, fournissant une résolution en paires de bases de la jonction des points de rupture. Cela est particulièrement précieux pour les CNV dans des régions répétitives—dupliqués segmentaires, région MHC, ensembles de gènes en tandem—où les signaux de profondeur des lectures courtes sont peu fiables. L'outil HiFiCNV de PacBio (2024) est le premier outil optimisé pour la détection des CNV à partir de lectures longues. Un benchmark de 2024 a révélé que les lectures HiFi à 15× détectaient environ 30 % de CNV en plus que le WGS à lectures courtes à 30× dans les mêmes échantillons, avec des appels supplémentaires concentrés dans les duplications segmentaires. Le compromis est le coût : le séquençage à lectures longues avec une couverture génomique équivalente est 3 à 5 fois plus cher que les méthodes à lectures courtes.
Figure 2 : Sensibilité de détection des CNV en fonction de la profondeur de séquençage et de la taille des CNV
Outils de bioinformatique pour la détection des CNV — Base algorithmique et performance de référence
Une étude de référence de 2024 dans Genome Biology a évalué six outils d'appel de CNV sur une lignée cellulaire cancéreuse hyper-diploïde (HCC1395) avec des données WGS et WES appariées, générant des données de performance exploitables pour la sélection des outils.
CNVkitConçu pour les données WES avec des échantillons tumoraux-normaux appariés. CNVkit construit une référence groupée à partir d'échantillons normaux, corrige le biais GC et segmente le signal de couverture en utilisant la segmentation binaire circulaire. C'est l'outil CNV WES le plus largement validé en génomique du cancer. Résultat de référence : >90 % de précision pour la détection des CNV somatiques WES avec des témoins normaux appariés. Idéal pour : analyse WES du cancer au niveau projet avec >10 échantillons normaux.
GATK gCNVDéveloppé pour la détection des CNV germinaux par séquençage du génome entier (WGS) à l'échelle de la population. Utilise un modèle bayésien avec segmentation PELT qui apprend le profil de couverture à partir d'une cohorte (ne nécessitant pas de contrôles normaux appariés pour chaque échantillon). Standard pour les grandes cohortes WGS (plus de 100 échantillons). Résultat de référence : meilleur rappel (>85%) pour les CNV germinaux rares grâce à un débruitage conscient de la cohorte. Idéal pour : études de CNV germinaux par WGS à grande échelle où aucun contrôle normal n'est disponible.
Control-FREECIdentifie les CNV en utilisant à la fois la profondeur de lecture et la fréquence de l'allèle B, permettant la détection en l'absence de contrôles normaux appariés. Sa capacité spécifique à l'allèle est utile pour les échantillons avec contamination de cellules normales : la fréquence de l'allèle B peut révéler des CNV là où la profondeur seule est ambiguë. Résultat de référence : précision modérée (~80 %) mais meilleure flexibilité à travers les types de données. Meilleur pour : projets avec des échantillons de contrôle limités ou lorsque des informations alléliques sont nécessaires.
Toile: Le détecteur de CNV recommandé par Illumina pour le WGS et le WES. Intègre la profondeur avec correction GC/mappabilité et BAF à partir des lectures SNP. Optimisé pour le pipeline Illumina DRAGEN. Résultat de référence : >90 % de précision pour les CNV somatiques WGS, comparable à CNVkit pour le WES. Meilleur pour : flux de travail uniquement Illumina et intégration de pipeline automatisée.
ÉCOLE: Appelant CNV WES basé sur l'apprentissage profond (réseau de neurones convolutif). Entraîné sur des données simulées avec du bruit spécifique à la capture, atteignant des taux de faux positifs inférieurs à ceux de CNVkit pour les CNV à exon unique. Résultat de référence : 20-30 % de faux positifs en moins que CNVkit pour WES. Meilleur pour : projets WES nécessitant une haute spécificité, en particulier lorsque la capacité de validation est limitée.
HiFiCNV: L'outil de détection des CNV à lecture longue de PacBio pour les lectures HiFi. Segmente la couverture des lectures longues après correction GC, tirant parti de la haute mappabilité des longues lectures. Outil en phase de développement. Résultat de référence : détecte environ 30 % de CNV en plus dans les régions répétitives par rapport aux méthodes à lecture courte. Idéal pour : les projets utilisant des données PacBio HiFi nécessitant la détection de CNV dans des régions génomiques complexes.
Services de détection de CNV soutenir chacun de ces outils avec des flux de travail validés, permettant aux chercheurs de sélectionner l'outil approprié en fonction de leur type de données et de leurs objectifs de projet.
Figure 3 : Pipeline bioinformatique CNV — étapes clés du traitement des lectures brutes aux appels de nombre de copies
Principaux défis qui compromettent la précision de la détection des CNV
La détection des CNV est plus sensible aux artefacts techniques que la détection des SNV, car la profondeur de lecture est affectée par plusieurs facteurs indépendants du nombre de copies biologique. Comprendre ces facteurs de confusion et appliquer des corrections appropriées est essentiel pour obtenir des résultats fiables.
biais GCL'efficacité de l'amplification PCR varie avec le contenu en GC sur une plage de 2 à 5 fois, créant une variation systématique de profondeur qui imite les signaux de CNV. La correction GC calcule les rapports de profondeur observée à attendue dans des fenêtres appariées en GC, mais cette correction est imparfaite pour les échantillons d'ADN fragmentés ou à faible entrée. Le biais résiduel après correction représente une fraction substantielle des faux positifs de CNV dans les données WGS et WES, en particulier dans les régions promoteurs riches en GC et les régions intergéniques pauvres en GC.
MappabilitéEnviron 10-15 % du génome humain—centromères, télomères, ensembles d'ADN ribosomal et duplications segmentaires—ne peuvent pas être cartographiés de manière unique par des lectures courtes et doivent être exclus de l'analyse des CNV. Les CNV dans ces régions sont systématiquement manqués. Pour le WES, la fraction inaccessible dépend de la conception du kit de capture et peut atteindre 15-20 % des régions ciblées.
Exigence normale correspondanteL'appel de CNV à échantillon unique - identifier les CNV à partir de la profondeur d'un échantillon sans comparaison de référence - a une précision limitée car la variation de profondeur technique ne peut pas être distinguée du signal CNV biologique en isolation. La pratique standard utilise soit un contrôle normal apparié (cancer somatique), soit une référence groupée provenant de ≥10 échantillons normaux (WGS germinal) ou ≥30 échantillons (WES germinal). Les projets respectant ces minimums utilisent des outils avec une normalisation basée sur un modèle intégrée (GATK gCNV) comme alternative.
artéfacts FFPEL'ADN FFPE a des tailles de fragments moyennes <300 pb et des bases déaminées dues au réticulation par le formaldéhyde. Ces propriétés augmentent la variance de profondeur et réduisent le rapport signal-bruit des CNV. Un benchmark de 2024 a révélé que la précision de détection des CNV diminue de 15 à 25 % pour l'FFPE par rapport aux tissus frais congelés. Les stratégies d'atténuation incluent des contrôles normaux FFPE appariés, une profondeur de séquençage accrue et des méthodes de normalisation spécialisées pour l'ADN fragmenté.
Pureté et hétérogénéité des tumeursDans les échantillons de cancer, le signal CNV effectif est le produit de l'état CNV, de la fraction de cellules tumorales portant ce CNV et de la pureté de l'échantillon. Un CNV présent dans 40 % des cellules tumorales dans un échantillon pur à 60 % produit un changement de profondeur de seulement 12 % par rapport au diploïde, indistinguable du bruit dans la plupart des pipelines. Les outils basés sur le BAF (Control-FREEC) maintiennent la détection à une pureté allant jusqu'à ~20 %, tandis que les méthodes basées uniquement sur la profondeur nécessitent >30 % pour l'appel de CNV somatiques.
Limites de faible couverture pour des types de CNV spécifiquesDifférents types de CNV ont des limites de détection différentes à la même profondeur. Les amplifications (gains) produisent des signaux plus faibles que les délétions — une triplication ne produit qu'une augmentation de 50 % de la profondeur attendue par rapport à la diminution de 50 % d'une délétions à copie unique. À 30× WGS, les deux sont détectables, mais à 1× LP-WGS, les amplifications nécessitent des événements 2-3× plus grands pour atteindre une confiance comparable. Les délétions homozygotes produisent le signal le plus fort (profondeur approchant zéro au niveau cible) et sont détectables même aux profondeurs les plus faibles. Comprendre ces limites de détection spécifiques aux types est important pour planifier des projets visant à détecter des classes spécifiques de CNV.
Figure 4 : Défis de l'analyse des CNV tumoraux — l'effet combiné de la pureté, de la qualité de l'échantillon et de la profondeur de séquençage

Analyse des CNV dans le cancer — Considérations somatiques vs germinales
La détection des CNV somatiques dans le cancer diffère de la détection des CNV germinaux sur trois points critiques : l'exigence d'un échantillon normal apparié, la présence de CNV subclonaux et l'effet perturbateur des changements de ploïdie tumorale.
Un échantillon normal apparié (sang ou tissu normal adjacent) est essentiel pour distinguer les CNV somatiques des CNV germinaux hérités et des artefacts techniques. Le normal apparié fournit un biais GC spécifique à l'échantillon, une efficacité de capture et une correction des effets de séquençage. En son absence, des références normales groupées peuvent compenser partiellement mais avec une sensibilité réduite. Pour les études sur le cancer, le LP-WGS à 1-2× est rapidement adopté pour le profilage des CNV somatiques dans de grandes cohortes - une étude de 2025 sur 2 000 échantillons de cancer a révélé que le LP-WGS à 1× détectait des amplifications focales et des délétions homozygotes avec plus de 85 % d'accord avec le WGS à 30× pour des événements supérieurs à 100 kb.
Les signatures de nombre de copies—des motifs génomiques d'amplifications et de délétions liés à des processus mutationnels spécifiques—fournissent des informations au-delà des altérations géniques individuelles. Le score HRD (déficience en recombinaison homologue) dérivé des motifs CNV à l'échelle du génome est établi comme un biomarqueur prédictif de la réponse à la thérapie par inhibiteurs de PARP. Services de WGS clinique prendre en charge à la fois des configurations à profondeur standard et à filtrage passe-bas pour l'analyse des CNV du cancer, avec des protocoles appariés tumeur-norme disponibles.
Détection de CNV à cellule uniqueUne frontière émergente dans l'analyse des CNV est le profilage des CNV à cellule unique, qui résout l'hétérogénéité intra-tumorale qui est moyennée dans le séquençage en vrac. Le séquençage génomique à cellule unique à faible profondeur (0,1-1× par cellule) combiné avec des algorithmes d'inférence de nombre de copies peut reconstruire l'architecture clonale d'une tumeur en détectant les différences de CNV entre les cellules individuelles. Cette approche a été utilisée pour retracer les modèles de dissémination métastatique et pour identifier des sous-clones rares portant des CNV conférant une résistance qui sont invisibles dans l'analyse en vrac. Le compromis est que les données à cellule unique présentent un bruit plus élevé par cellule que les données en vrac, nécessitant des algorithmes de normalisation et d'appel spécialisés ainsi qu'un plus grand nombre de cellules pour une attribution confiante des CNV. Les projets envisageant l'analyse des CNV à cellule unique devraient prévoir au moins 100 à 500 cellules par échantillon pour atteindre une puissance statistique suffisante pour la détection des sous-clones.
Figure 5 : Guide de sélection des outils d'analyse CNV — associer l'outil au type de données et à l'objectif de recherche

Séquençage Long-Read pour la Détection des CNV
Le séquençage à lecture longue répond à la limitation fondamentale de la détection des CNV par lectures courtes : l'incapacité à mapper les lectures dans des régions répétitives et l'incapacité à couvrir les points de rupture. Les lectures PacBio HiFi de 10 à 20 kb avec une précision de plus de 99,9 % peuvent directement couvrir les points de rupture des CNV, tandis que les lectures ultra-longues Nanopore dépassant 100 kb peuvent couvrir des réarrangements complexes entiers.
Un benchmark de 2024 sur la détection des CNV à lecture longue a révélé que les lectures HiFi à 15× détectaient environ 30 % de CNV de plus que le séquençage génomique à courte lecture (WGS) à 30× dans les mêmes échantillons, avec des appels supplémentaires concentrés dans les duplications segmentaires et d'autres régions répétitives. La résolution des points de rupture s'est améliorée, passant de 1-10 kb (courte lecture) à moins de 100 pb (HiFi).
La détection des CNV longs est particulièrement précieuse lorsque l'accent de la recherche porte sur des régions génomiques complexes : duplications segmentaires, région MHC, familles de gènes dupliqués en tandem ou zones de CNV connues. Pour les projets axés sur plus de 90 % du génome accessible aux lectures courtes, les méthodes de lectures courtes restent le choix pratique en raison de leur coût inférieur et de leurs outils plus matures.
Stratégies hybrides pour une analyse complète des CNVPour les projets nécessitant à la fois un dépistage génomique à grande échelle rentable et une analyse de points de rupture à haute résolution, une approche hybride combinant le séquençage génomique à lecture courte (LP-WGS) à 1-2× pour l'identification initiale des CNV avec le séquençage ciblé à lecture longue des régions de points de rupture des CNV offre la meilleure utilisation des ressources. Le dépistage LP-WGS identifie les CNV candidats et estime leurs limites approximatives, tandis que le séquençage à lecture longue résout le point de rupture précis et identifie l'architecture séquentielle sous-jacente (par exemple, NAHR entre des éléments répétés spécifiques). Cette approche par étapes a été utilisée avec succès dans des pipelines de validation clinique des CNV et dans des études de population où une caractérisation complète des CNV est nécessaire, mais où les contraintes budgétaires limitent l'utilisation du séquençage génomique à lecture longue.
Validation CNV — Le Rôle des Méthodes Orthogonales
Les appels de CNV computationnels doivent être validés par des méthodes orthogonales avant de tirer des conclusions biologiques solides. La PCR digitale en gouttelettes (ddPCR) fournit une quantification absolue du nombre de copies à des loci spécifiques et est pratique pour valider 5 à 20 candidats par projet, avec une sensibilité de détection suffisante pour confirmer des gains ou des pertes de copies uniques dans des échantillons avec 50 % ou plus de pureté tumorale. Le microarray chromosomique (aCGH) reste la référence mondiale avec une sensibilité et une spécificité supérieures à 95 % pour les CNV > 50 kb, et il sert de plateforme de référence pour la plupart des pipelines de validation clinique des CNV. Pour les projets rapportant des CNV associés à des maladies nouvelles ou des résultats de qualité clinique, la validation par au moins une méthode orthogonale est une pratique standard avant publication ou rapport clinique.
Bases de données CNV pour l'annotation et l'interprétation
Navigateur de génome UCSCPlateforme de visualisation principale pour les appels CNV dans un contexte génomique, avec des pistes pour les éléments répétés, les gènes connus, les duplications segmentaires et la fréquence des CNV dans la population provenant de 1000 Genomes et gnomAD.
Base de données DECIPHER: Curate les CNVs associés aux troubles génétiques, liant chaque variante aux phénotypes cliniques et au contenu génétique pour l'évaluation de la pathogénicité.
Base de données ClinGenLes scores de sensibilité de dosage au niveau des gènes—évaluations systématiques de l'haploinsuffisance et de la triplosensibilité pour chaque gène—guident l'interprétation de la probabilité qu'un CNV contenant un gène soit pathogène. Services d'analyse de données génomiques intégrer ces bases de données dans des flux de travail d'annotation automatisés.
Ressources informatiques pour l'analyse des CNV
L'analyse CNV est modeste sur le plan computationnel par rapport à l'alignement WGS ou à l'assemblage de novo. Un échantillon WGS à 30× nécessite environ 1 à 2 heures pour l'appel CNV (après l'alignement) sur un nœud de calcul standard. L'appel CNV WES avec CNVkit nécessite 30 à 60 minutes par échantillon. Le LP-WGS à 1× se traite en 15 à 30 minutes. GATK gCNV nécessite un traitement au niveau de la cohorte — après que le modèle est construit à partir de la cohorte (1 à 2 heures), l'appel d'échantillons individuels est rapide (10 à 20 minutes par échantillon). Les exigences de stockage sont dominées par les fichiers BAM/CRAM alignés nécessaires pour l'extraction de profondeur. Les projets utilisant l'informatique en nuage devraient budgétiser les ressources de calcul en fonction du nombre d'échantillons et du volume de données.
Figure 6 : Feuille de route du projet d'analyse CNV — de la question de recherche à l'interprétation des CNV
FAQ
Quelle profondeur de séquençage est nécessaire pour la détection des CNV à partir du séquençage du génome entier ?
Pour la détection des CNV de plus de 5-10 kb dans tout le génome, une couverture de 15-30× est standard. Le séquençage génomique à faible profondeur (LP-WGS) à 1-2× détecte les CNV de plus de 50-100 kb pour le dépistage clinique où l'efficacité des coûts est priorisée.
Comment la qualité des échantillons FFPE affecte-t-elle la détection des CNV ?
Les échantillons FFPE contiennent de l'ADN fragmenté et des dommages à la base qui augmentent le bruit de profondeur de lecture, réduisant la sensibilité de détection des CNV de 15 à 25 % par rapport aux tissus frais congelés. L'utilisation de contrôles normaux FFPE appariés et l'augmentation de la profondeur de séquençage compensent partiellement cela.
Quelle est la pureté tumorale minimale pour la détection des CNV somatiques ?
La plupart des outils nécessitent une pureté supérieure à 20-30 % pour une détection fiable. Les outils basés sur BAF (Control-FREEC) fonctionnent mieux à faible pureté que les méthodes basées uniquement sur la profondeur.
Comment choisir entre CNVkit et GATK gCNV ?
GATK gCNV est recommandé pour la détection de CNV germinaux à grande échelle dans le séquençage génomique entier (WGS) où un modèle de population peut être construit. CNVkit est recommandé pour les CNV dans le séquençage de l'exome (WES) au niveau du projet avec des contrôles normaux appariés.
Puis-je détecter des CNV à partir de données RNA-seq ?
La détection des CNV par RNA-seq est possible mais moins fiable que les méthodes basées sur l'ADN en raison de la variation d'expression. Il est recommandé de valider toute découverte de CNV par RNA-seq par des méthodes basées sur l'ADN.
Quelle est la taille de référence minimale pour l'analyse CNV à normalisation groupée ?
Minimum 10 échantillons normaux pour le WGS et 30 pour le WES, reflétant le niveau de bruit plus élevé de la capture ciblée. En dessous de ces seuils, les méthodes basées sur des modèles (GATK gCNV) sont préférées.
Références
- Évaluation de la détection des variations du nombre de copies somatiques par les technologies NGS et les outils bioinformatiques sur un génome cancéreux hyper-diploïde. Genome Biology. 2024;25:164.
- ECOLE : Apprendre à identifier les variants de nombre de copies sur des données de séquençage de tout l'exome. Nature Communications. 2023 ; 14 : 44116.
- Évaluation de la détection des variations du nombre de copies avec le séquençage du génome entier à faible couverture. Briefings in Bioinformatics. 2025;26:bbaf514.
- Long et précis : Comment le séquençage HiFi transforme la recherche génomique. Génomique, Protéomique et Bioinformatique. 2025 ;23:qzaf003.
- Le séquençage du génome entier à faible débit est une méthode fiable et économique pour la détection des CNV. Annales de Génétique Humaine. 2023;87:251-260.
Services connexes
- Séquençage du génome entier
- Séquençage de l'exome complet
- Appel de variants
- Séquençage de nouvelle génération
- Analyse des données génomiques
- Services de bioinformatique
- Services WGS
- Services WES
À des fins de recherche uniquement, non destiné à un diagnostic clinique, un traitement ou des évaluations de santé individuelles.


